Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Tingdong Li and Version 4 by Camila Xu.
Herpesviruses are major pathogens that infect humans and animals. Manipulating the large genome is critical for exploring the function of specific genes and studying the pathogenesis of herpesviruses and developing novel anti-viral vaccines and therapeutics. Bacterial artificial chromosome (BAC) technology significantly advanced the capacity of herpesviruses researchers to manipulate the virus genomes.
- herpesvirus
- bacterial artificial chromosomes
- gene editing
- virus mutant selection
1. Introduction
Genome manipulation is an effective method for studying the function of viral genes and can help scientists understand the biology of viruses, such as discovering virulence factors and exploring new targets for developing novel vaccines and antiviral drugs for the prevention and treatment of viral infection. Herpesviruses are a group of double-strand DNA (dsDNA) viruses which can cause lifelong persistent infections and are major pathogens in humans and a wide range of animals. The genomes of herpesviruses are between 125 and 295 kilobases in length [1], which makes it difficult for genome manipulation.
In the past decades, scientists have developed several methods for manipulating the genome of herpesviruses, such as λ-red, Cre-loxp, CRISPR-Cas9, and others, which helped understand the pathogenesis of herpesviruses and develop novel vaccines and antiviral drugs [2][3][2,3]. Traditionally, linear DNA fragments or circular plasmids containing selection cassettes and flanking homologous regions were transferred into herpesvirus-infected cells, and the genomes of herpesviruses were edited by recombination, and then the recombinant viruses were selected by using different markers (different selection cassettes) [4][5][6][7][4,5,6,7 ]. This approach has a low efficiency. In addition, its dependence on the production of progeny viruses results in a limited range of genes that can be edited. Moreover, the cumbersome screening process for identifying functional recombinant viruses restricts its application [3]. Therefore, it is imperative to explore alternative techniques that can enhance the efficiency and expand the range of genes that can be edited. Bacterial artificial chromosomes (BAC), constructed based on factor F of Escherichia coli (E. coli), provide an important technical platform for the editing of large genomes, including herpesviruses [8][9][10][11][8,9,10,11]. BAC-based gene editing strategies are independent of the production of the progeny, and the screening efficiency is higher than conventional strategies [12][13][14][12,13,14]. Firstly, BACs can accommodate the insertion and stable inheritance of exogenous gene fragments ranging from 100 to 300 kb. The F factor facilitates the maintenance of low copy numbers (1–2 copies per cell) in E. coli which greatly reduces the difficulty in screening. Meanwhile, the development of various gene editing techniques in E. coli has greatly enhanced the editing efficiency of herpesvirus BACs compared to traditional methods. Edited BACs can be reintroduced into eukaryotic cells to reconstruct herpesvirus mutants for further study of their biological behaviors in cells. In addition, the selection of recombinantly engineered viruses is not dependent on the generation of progeny viruses, allowing editing of the γ herpes virus genome that maintains latent infection in the long term [13][15][13,15] as well as manipulating genes essential for the production of progeny viruses [11].
2. Genome Editing Techniques of Herpesviruses Based on BAC
2.1. RecA Recombination Technique
The RecA recombination technique is a bacterial endogenous homologous recombination system composed of RecA recombinase and related auxiliary proteins (such as RecBCD, RecFOR, RuvABC, etc.). RecBCD is a multifunctional enzyme complex consisting of RecB, RecC, and RecD. When binding to the incision on the double-stranded donor DNA, RecB and RecD exhibit 3′ to 5′ and 5′ to 3′ helicase activities, respectively, which together result in the opening of the complementary double-stranded DNA (Figure 1a) [16]. Then, RecC can mediate the recognition of chi sites in the sequence and thereby cause allosteric changes, leading to a decrease in the pace of movement (Figure 1b) [17]. At this point, RecB also has exonuclease activity, which mediates the generation of single-stranded DNA at the 3′ end (Figure 1c) [18][19][20][18,19,20]. The RecA recombinase binds to single-stranded DNA sites generated by the exonucleases and engages in the process of identifying homologous regions on the BAC for subsequent annealing and interaction. After successful interaction, the nucleoprotein filaments invade the dsDNA and undergo strand exchange to form heteroduplex DNA (Figure 1d). Finally, RuvABC assists in catalyzing branch migration and degradation of the heteroduplex DNA, resulting in homologous recombination (Figure 1e) [21].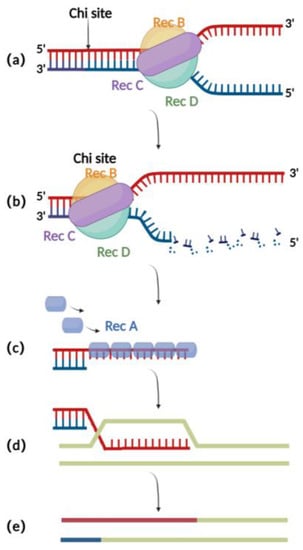
Figure 1 . Schematic diagram of RecA recombination technique:(a)RecBCD binds to the double-stranded DNA terminus and promotes unwinding;(b)At the chi site, a single-stranded DNA is produced at the 3′ end;(c) The RecA protein binds to single-stranded DNA at the 3′ end;(d)ssDNA-RecA invades intact homologous double-stranded DNA for strand exchange;(e) Recombination completed.
Table 1.
The pros and cons of BAC-based herpesvirus genome editing techniques.
| Editing Techniques | Length of Homologous Arms | Editing Efficiency | Requirement for Editing Site | Stability of BAC |
|---|---|---|---|---|
| RecA Recombination | 500 bp–3 k bp | 10−6 to 10−4 | None | Causes mutations in the BAC |
| λ-red Recombination | 30–50 bp | <1% | None | Maintained the stability of BAC |
| Base Editing | Not Required | Approaching 100% | PAM Sites & Base Editing Sites; Only Base Editing can be done | Maintained the stability of BAC |
2.2. λ-Red Recombination Technique
Homologous recombination mediated by the λ-red recombination system is currently the most widely used recombination technique for editing herpesvirus BAC clones [2][25][2,25]. The λ-red recombination system is derived from the phage λ and consists of Gam, Exo, and Beta proteins. The Gam protein is an auxiliary protein of Exo protein and Beta protein. The Gam protein can inhibit the binding of Rec BCD to the ends of dsDNA and thus inhibit the function of Rec BCD exonuclease, preventing the degradation of the exogenous double-stranded DNA (Figure 2a) [26][27][26,27]. Exo proteins can bind to the ends of a dsDNA donor, which contains homologous fragments on both sides of the target gene. Concurrently, Exo protein possesses 5′ to 3′ exonuclease activity, producing a stretch of single-stranded DNA at the 3′ end (Figure 2b) [28]. Beta protein plays a decisive role in the process of λ-red homologous recombination. As a single-stranded DNA binding protein, the beta protein binds to the single-stranded DNA produced by the Exo protein. The binding of Beta protein enhances the annealing of the donor DNA fragment and the homologous sequence at the target site of the replicating herpes virus BAC (Figure 2c). Homologous recombination is complete with the replication of DNA (Figure 2d) [29][30][29,30].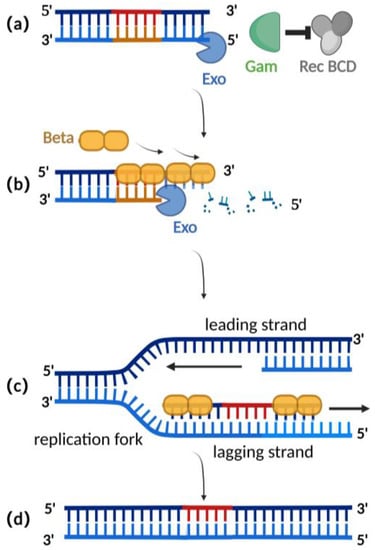
Figure 2. Schematic diagram of λ-red recombination technique: (a) Gam protein inhibits the activity of Rec BCD exonuclease; (b) Exo protein creates a stretch of single-stranded DNA at the 3′ end; (c) Beta proteins bind here, facilitating annealing interactions between the donor DNA fragment and the homologous sequence of the target site; (d) homologous recombination is complete with the replication of DNA.
2.3. Base Editing Technique
CRISPR/Cas 9 is a powerful genome editing technique [36] that has been successfully applied in a wide range of eukaryotic cells, including human cell lines, embryonic stem cells, mice, Arabidopsis, and Drosophila [37][38][39][40][37,38,39,40]. In 2013, Jiang et al. successfully edited Streptococcus pneumonia’s genome using CRISPR/Cas-9-only gene editing [41]. Since then, it has been successfully employed in a variety of prokaryotic species as well [42][43][42,43]. The versatility and effectiveness of CRISPR/Cas9 in modifying the genome make it a valuable tool for a wide range of scientific and medical applications. Due to the lack of the non-homologous end-joining (NHEJ) pathway [44][45][46][47][44,45,46,47] and the low efficiency of their endogenous homologous recombination system, it is difficult to achieve stable genome editing in most bacteria using CRISPR/Cas9 gene editing technology alone [41][48][49][50][41,48,49,50]. Until now, there was still no publication reporting CRISPR/Cas 9-based gene editing technique in the stably preserved herpesvirus BAC gene in E. coli. This highlights the need to explore alternative approaches to achieve efficient and reliable gene editing in these organisms. Recently, scientists have constructed an efficient gene editing method by combing CRISPR/Cas 9 gene editing technique with precise base editing technology. This method consists of an sgRNA and a complex that includes modified Cas9 proteins, cytosine deaminases, and an uracil glycosylase inhibitor (UGI) [51]. Unlike wild-type Cas9 proteins, the modified Cas9 is catalytically dead, lacking endonuclease activity. Therefore, it can only facilitate genome targeting via sgRNA but cannot induce a double strand break (DSB) due to the absence of cleavage activity (Figure 3a). Cytosine deaminase converts the specified cytosine (C) site to uracil (U) (Figure 3b). At this point, uracil glycosylase inhibitors can prevent the excision of intermediate product U, increasing the efficiency of converting C to T on the DNA chain, ultimately achieving single-base precise editing of C to T and G to A (Figure 3c).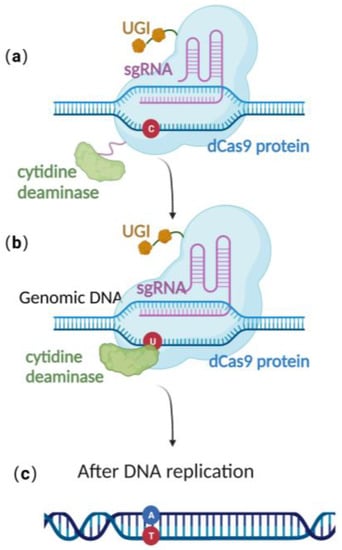
Figure 3. Schematic diagram of base editing method: (a) dCas9 mediates targeting without DSB formation; (b) cytosine deaminase converts C to U; (c) single-base precise editing is complete with the replication of DNA.