Mitochondrial open reading frame of the 12S rRNA type-c (MOTS-c) is the most unearthed peptide encoded by mitochondrial DNA (mtDNA). It is an important regulator of the nuclear genome during times of stress because it promotes an adaptive stress response to maintain cellular homeostasis. Identifying MOTS-c specific binding partners may aid in deciphering the complex web of mitochondrial and nuclear-encoded signals. Mitochondrial damage and dysfunction have been linked to aging and the accelerated cell death associated with many types of retinal degenerations. Furthermore, research on MOTS-c ability to revive oxidatively stressed RPE cells has revealed a significant protective role for the molecule. Evidence suggests that senescent cells play a role in the development of age-related retinal disorders.
- MOTS-c
- glaucoma
- RPE
1. Introduction
2. Age-Related Retinal Diseases
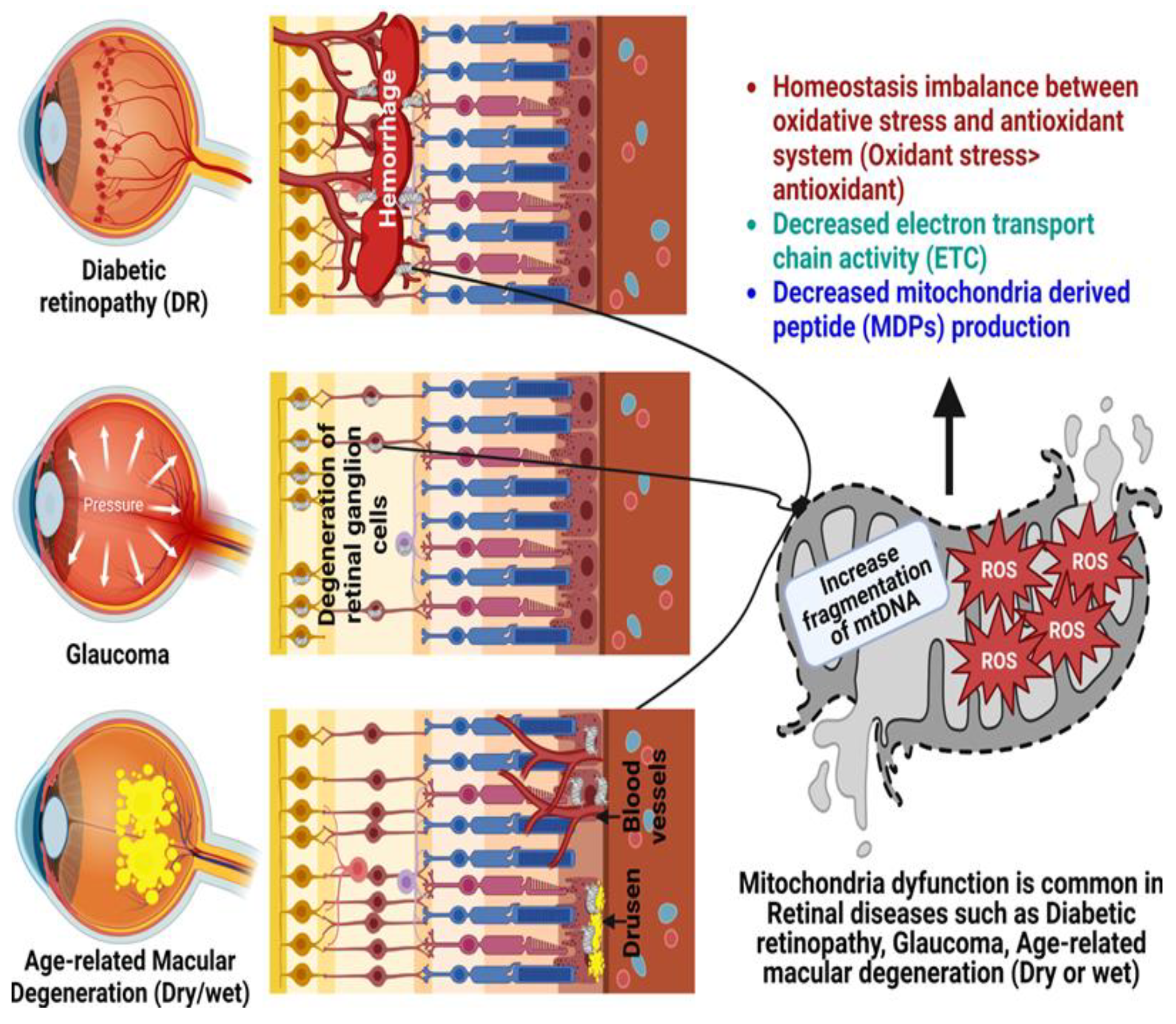
2.1. Glaucoma
2.2. Diabetic Retinopathy
2.3. Age-Related Macular Degeneration
References
- Cobb, L.J.; Lee, C.; Xiao, J.; Yen, K.; Wong, R.G.; Nakamura, H.K.; Mehta, H.H.; Gao, Q.; Ashur, C.; Huffman, D.M.; et al. Naturally occurring mitochondrial-derived peptides are age-dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 2016, 8, 796–809.
- Kim, S.J.; Xiao, J.; Wan, J.; Cohen, P.; Yen, K. Mitochondrially derived peptides as novel regulators of metabolism. J. Physiol. 2017, 595, 6613–6621.
- Kim, S.J.; Miller, B.; Kumagai, H.; Silverstein, A.R.; Flores, M.; Yen, K. Mitochondrial-derived peptides in aging and age-related diseases. Geroscience 2021, 43, 1113–1121.
- Mohtashami, Z.; Singh, M.K.; Salimiaghdam, N.; Ozgul, M.; Kenney, M.C. MOTS-c, the Most Recent Mitochondrial Derived Peptide in Human Aging and Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 1991.
- Sreekumar, P.G.; Kannan, R. Mechanisms of protection of retinal pigment epithelial cells from oxidant injury by humanin and other mitochondrial-derived peptides: Implications for age-related macular degeneration. Redox Biol. 2020, 37, 101663.
- Wu, Y.; Sun, L.; Zhuang, Z.; Hu, X.; Dong, D. Mitochondrial-Derived Peptides in Diabetes and Its Complications. Front. Endocrinol. 2021, 12, 808120.
- Nashine, S.; Kenney, M.C. Effects of Mitochondrial-Derived Peptides (MDPs) on Mitochondrial and Cellular Health in AMD. Cells 2020, 9, 1102.
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403.
- Carrella, S.; Massa, F.; Indrieri, A. The Role of MicroRNAs in Mitochondria-Mediated Eye Diseases. Front. Cell Dev. Biol. 2021, 9, 653522.
- Schrier, S.A.; Falk, M.J. Mitochondrial disorders and the eye. Curr. Opin. Ophthalmol. 2011, 22, 325–331.
- Bourne, R.; Steinmetz, J.D.; Flaxman, S.; Briant, P.S.; Taylor, H.R.; Resnikoff, S.; Tareque, M.I. Trends in prevalence of blindness and distance and near vision impairment over 30 years: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e130–e143.
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40.
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014.
- Cavallotti, C.; Artico, M.; Pescosolido, N.; Leali, F.M.; Feher, J. Age-related changes in the human retina. Can. J. Ophthalmol. 2004, 39, 61–68.
- Samuel, M.A.; Zhang, Y.; Meister, M.; Sanes, J.R. Age-related alterations in neurons of the mouse retina. J. Neurosci. 2011, 31, 16033–16044.
- Blasiak, J.; Sobczuk, P.; Pawlowska, E.; Kaarniranta, K. Interplay between aging and other factors of the pathogenesis of age-related macular degeneration. Ageing Res. Rev. 2022, 81, 101735.
- Jarrett, S.G.; Lewin, A.S.; Boulton, M.E. The importance of mitochondria in age-related and inherited eye disorders. Ophthalmic Res. 2010, 44, 179–190.
- Gupta, N.; Yücel, Y.H. Glaucoma as a neurodegenerative disease. Curr. Opin. Ophthalmol. 2007, 18, 110–114.
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23.
- Deliyanti, D.; Alrashdi, S.F.; Tan, S.M.; Meyer, C.; Ward, K.W.; de Haan, J.B.; Wilkinson-Berka, J.L. Nrf2 Activation Is a Potential Therapeutic Approach to Attenuate Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 815–825.
- Gauthier, A.C.; Liu, J. Neurodegeneration and Neuroprotection in Glaucoma. Yale J. Biol. Med. 2016, 89, 73–79.
- Agarwal, R.; Gupta, S.K.; Agarwal, P.; Saxena, R.; Agrawal, S.S. Current concepts in the pathophysiology of glaucoma. Indian J. Ophthalmol. 2009, 57, 257–266.
- Saccà, S.C.; Izzotti, A. Oxidative stress and glaucoma: Injury in the anterior segment of the eye. Prog. Brain Res. 2008, 173, 385–407.
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541.
- Lo Faro, V.; Nolte, I.M.; Ten Brink, J.B.; Snieder, H.; Jansonius, N.M.; Bergen, A.A. Mitochondrial Genome Study Identifies Association Between Primary Open-Angle Glaucoma and Variants in MT-CYB, MT-ND4 Genes and Haplogroups. Front. Genet. 2021, 12, 781189.
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Jonasdottir, A.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874.
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909.
- Aslan, M.; Dogan, S.; Kucuksayan, E. Oxidative stress and potential applications of free radical scavengers in glaucoma. Redox Rep. 2013, 18, 76–87.
- Adornetto, A.; Rombolà, L.; Morrone, L.A.; Nucci, C.; Corasaniti, M.T.; Bagetta, G.; Russo, R. Natural Products: Evidence for Neuroprotection to Be Exploited in Glaucoma. Nutrients 2020, 12, 3158.
- Oikawa, K.; Ver Hoeve, J.N.; Teixeira, L.B.C.; Snyder, K.C.; Kiland, J.A.; Ellinwood, N.M.; McLellan, G.J. Sub-region-Specific Optic Nerve Head Glial Activation in Glaucoma. Mol. Neurobiol. 2020, 57, 2620–2638.
- Sapienza, A.; Raveu, A.L.; Reboussin, E.; Roubeix, C.; Boucher, C.; Dégardin, J.; Godefroy, D.; Rostène, W.; Reaux-Le Goazigo, A.; Baudouin, C.; et al. Bilateral neuroinflammatory processes in visual pathways induced by unilateral ocular hypertension in the rat. J. Neuroinflamm. 2016, 13, 44.
- Nita, M.; Grzybowski, A. The Role of the Reactive Oxygen Species and Oxidative Stress in the Pathomechanism of the Age-Related Ocular Diseases and Other Pathologies of the Anterior and Posterior Eye Segments in Adults. Oxid. Med. Cell. Longev. 2016, 2016, 3164734.
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513.
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777.
- Goodfellow, M.J.; Borcar, A.; Proctor, J.L.; Greco, T.; Rosenthal, R.E.; Fiskum, G. Transcriptional activation of antioxidant gene expression by Nrf2 protects against mitochondrial dysfunction and neuronal death associated with acute and chronic neurodegeneration. Exp. Neurol. 2020, 328, 113247.
- Guo, X.; Dason, E.S.; Zanon-Moreno, V.; Jiang, Q.; Nahirnyj, A.; Chan, D.; Flanagan, J.G.; Sivak, J.M. PGC-1α signaling coordinates susceptibility to metabolic and oxidative injury in the inner retina. Am. J. Pathol. 2014, 184, 1017–1029.
- Zuo, L.; Khan, R.S.; Lee, V.; Dine, K.; Wu, W.; Shindler, K.S. SIRT1 promotes RGC survival and delays loss of function following optic nerve crush. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5097–5102.
- Balaiya, S.; Abu-Amero, K.K.; Kondkar, A.A.; Chalam, K.V. Sirtuins Expression and Their Role in Retinal Diseases. Oxid. Med. Cell. Longev. 2017, 2017, 3187594.
- Balaiya, S.; Khetpal, V.; Chalam, K.V. Hypoxia initiates sirtuin1-mediated vascular endothelial growth factor activation in choroidal endothelial cells through hypoxia inducible factor-2α. Mol. Vis. 2012, 18, 114–120.
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154.
- Sykiotis, G.P. Keap1/Nrf2 Signaling Pathway. Antioxidants 2021, 10, 828.
- Ren, J.; Zhang, S.; Pan, Y.; Jin, M.; Li, J.; Luo, Y.; Sun, X.; Li, G. Diabetic retinopathy: Involved cells, biomarkers, and treatments. Front. Pharmacol. 2022, 13, 953691.
- Nian, S.; Lo, A.C.Y.; Mi, Y.; Ren, K.; Yang, D. Neurovascular unit in diabetic retinopathy: Pathophysiological roles and potential therapeutical targets. Eye Vis. 2021, 8, 15.
- Kenney, M.; Falatoonzadeh, P.; Atilano, S.; Chwa, M.; Caceres-del-Carpio, J.; Malik, D.; Kuppermann, B. African-origin mitochondrial DNA variants as a contributing factor to susceptibilities for diabetes and age-related diseases. Int. J. Diabetes Clin. Res. 2016, 3, 53.
- Dolinko, A.H.; Chwa, M.; Atilano, S.R.; Kenney, M.C. African and Asian Mitochondrial DNA Haplogroups Confer Resistance Against Diabetic Stresses on Retinal Pigment Epithelial Cybrid Cells In Vitro. Mol. Neurobiol. 2020, 57, 1636–1655.
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys. Ther. 2008, 88, 1322–1335.
- Nentwich, M.M.; Ulbig, M.W. Diabetic retinopathy—Ocular complications of diabetes mellitus. World J. Diabetes 2015, 6, 489–499.
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic. Biol. Med. 2003, 35, 1491–1499.
- Miller, D.J.; Cascio, M.A.; Rosca, M.G. Diabetic Retinopathy: The Role of Mitochondria in the Neural Retina and Microvascular Disease. Antioxidants 2020, 9, 905.
- Kowluru, R.A.; Chan, P.S. Oxidative stress and diabetic retinopathy. Exp. Diabetes Res. 2007, 2007, 43603.
- Trudeau, K.; Molina, A.J.; Guo, W.; Roy, S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: Implications for diabetic retinopathy. Am. J. Pathol. 2010, 177, 447–455.
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023.
- Romeo, G.; Liu, W.H.; Asnaghi, V.; Kern, T.S.; Lorenzi, M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes 2002, 51, 2241–2248.
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal. Transduct. 2012, 2012, 646354.
- Santos, J.M.; Mohammad, G.; Zhong, Q.; Kowluru, R.A. Diabetic retinopathy, superoxide damage and antioxidants. Curr. Pharm. Biotechnol. 2011, 12, 352–361.
- Johansen, J.S.; Harris, A.K.; Rychly, D.J.; Ergul, A. Oxidative stress and the use of antioxidants in diabetes: Linking basic science to clinical practice. Cardiovasc. Diabetol. 2005, 4, 5.
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Kanwar, M.; Chan, P.S.; Kern, T.S.; Kowluru, R.A. Oxidative damage in the retinal mitochondria of diabetic mice: Possible protection by superoxide dismutase. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3805–3811.
- Kim, S.J.; Miller, B.; Mehta, H.H.; Xiao, J.; Wan, J.; Arpawong, T.E.; Yen, K.; Cohen, P. The mitochondrial-derived peptide MOTS-c is a regulator of plasma metabolites and enhances insulin sensitivity. Physiol. Rep. 2019, 7, e14171.
- Ramanjaneya, M.; Bettahi, I.; Jerobin, J.; Chandra, P.; Abi Khalil, C.; Skarulis, M.; Atkin, S.L.; Abou-Samra, A.B. Mitochondrial-Derived Peptides Are Down Regulated in Diabetes Subjects. Front. Endocrinol. 2019, 10, 331.
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-related macular degeneration: Genetics and biology coming together. Annu. Rev. Genomics Hum. Genet. 2014, 15, 151–171.
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Aspects Med. 2012, 33, 295–317.
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Prim. 2021, 7, 31.
- Ding, X.; Patel, M.; Chan, C.C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18.
- Somasundaran, S.; Constable, I.J.; Mellough, C.B.; Carvalho, L.S. Retinal pigment epithelium and age-related macular degeneration: A review of major disease mechanisms. Clin. Exp. Ophthalmol. 2020, 48, 1043–1056.
- Datta, S.; Cano, M.; Ebrahimi, K.; Wang, L.; Handa, J.T. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog. Retin. Eye Res. 2017, 60, 201–218.
- Ferrington, D.A.; Fisher, C.R.; Kowluru, R.A. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol. Med. 2020, 26, 105–118.
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2848–2855.
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020, 79, 100858.
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467.
- Bohr, V.A.; Stevnsner, T.; de Souza-Pinto, N.C. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene 2002, 286, 127–134.
- Santos, J.H.; Meyer, J.N.; Skorvaga, M.; Annab, L.A.; Van Houten, B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell 2004, 3, 399–411.
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.T.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12.
- Toma, C.; De Cillà, S.; Palumbo, A.; Garhwal, D.P.; Grossini, E. Oxidative and Nitrosative Stress in Age-Related Macular Degeneration: A Review of Their Role in Different Stages of Disease. Antioxidants 2021, 10, 653.
- Tan, W.; Zou, J.; Yoshida, S.; Jiang, B.; Zhou, Y. The Role of Inflammation in Age-Related Macular Degeneration. Int. J. Biol. Sci. 2020, 16, 2989–3001.
- Khan, A.H.; Pierce, C.O.; De Salvo, G.; Griffiths, H.; Nelson, M.; Cree, A.J.; Menon, G.; Lotery, A.J. The effect of systemic levels of TNF-alpha and complement pathway activity on outcomes of VEGF inhibition in neovascular AMD. Eye 2021, 36, 2192–2199.
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464.
- Sreekumar, P.G.; Hinton, D.R.; Kannan, R. Endoplasmic reticulum-mitochondrial crosstalk: A novel role for the mitochondrial peptide humanin. Neural Regen. Res. 2017, 12, 35–38.
- Nashine, S.; Cohen, P.; Wan, J.; Kenney, M.C. Effect of Humanin G (HNG) on inflammation in age-related macular degeneration (AMD). Aging 2022, 14, 4247–4269.
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell. Metab. 2018, 28, 516–524.e7.