1. Introduction
Mitochondrial DNA (mtDNA) encodes for numerous small polypeptides, termed mitochondrial-derived peptide (MDP), from their Short Open Reading Frame regions. These MDPs include humanin (HN), six small-humanin like peptides (SHLP1-6), and
mitochondrial open reading frame of the 12S rRNA type-c (MOTS-c
) [1]. MDPs have been shown to reverse insulin resistance, slow the aging process, and reduce inflammation in rodent studies
[2][3][4][2,3,4]. They are known to provide a wide array of protective effects, including neuroprotective, cytoprotective, antioxidant, anti-inflammatory, and metabolic properties which would be valuable in the treatment of retinal diseases
[5][6][5,6]. Therefore, it is not surprising that all MDPs discovered to date have either altered expression under age-related pathological conditions or can ameliorate symptoms when administered exogenously.
The MDP that has been evaluated the most is HN, which is cytoprotective in numerous neurodegenerative diseases and shows anti-apoptotic activity in cybrid cell lines carrying mitochondria from patients with age-related macular degeneration (AMD)
[5]. Importantly, Nashine et al., showed that damaged mitochondria in the AMD patients be rescued by treatment with Humanin-G, a cytoprotective peptide that increased cellular longevity
[7]. The rate of damage to mtDNA is higher than that to nuclear DNA in studies with human RPE cells exposed to oxidative stress
[8]. Six other peptides, designated SHLP1-6, with lengths ranging from 20 to 38 amino acids, are also located on the 16S rRNA, alongside humanin. SHLP6 induced cell death, while SHLP2 and SHLP3 prevented cell death. SHLP2 acted similarly to HN in protecting primary mouse cortical neurons from toxicity caused by beta-amyloid (Aβ
1-42). The differentiation of insulin-dependent adipocytes was hastened by SHLP2 and SHLP3
[1].
Mitochondria are found in the retinal pigment epithelial (RPE) basal region, near the photoreceptors (PRs). In the inner retina, however, mitochondria are predominantly concentrated in the unmyelinated proximal axons of retinal ganglion cells (RGCs), which transmit visual information to the brain
[9]. Over time, oxidative damage caused by mtDNA instability leads to more damage to the mitochondria, which is known to be a major cause of age-related eye disorders
[10].
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are two types of free radicals that can be produced as metabolic byproducts of oxidation-reduction reactions in living cells
[11]. Unstable molecules with an odd number of unpaired electrons are known as free radicals. It is important to note that both endogenous and exogenous substances can contribute to ROS production
[11]. High ROS can react with a wide variety of biomolecules (such as proteins, lipid membranes, and DNA) and causing harm to cells. High level of ROS and free radicals, or low levels of antioxidants, characterize oxidative stress
[12][13][12,13]. Dynamic redox balance between ROS production and free radical scavenging may be achieved under physiological conditions. Massive accumulation of ROS occurs in response to different overabundant stimuli (both endogenous and exogenous). This may cause oxidative stress in the affected tissues
[14].
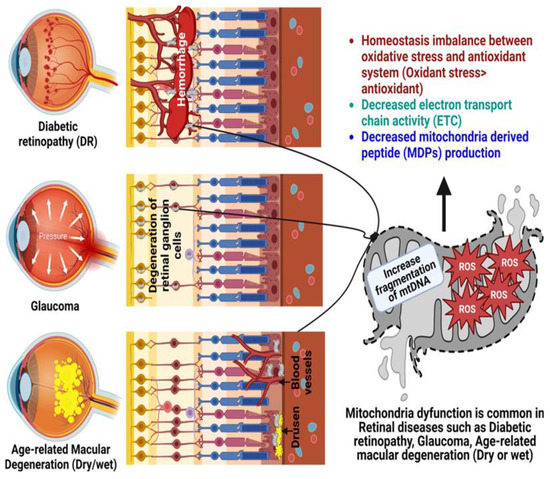
2. Age-Related Retinal Diseases
Metabolic dysfunction in neuronal cells is associated with the development of age-related neurodegenerative diseases. The net result of metabolic dysfunction includes reduced bioenergetics, increased generation of mitochondrial ROS, mitochondrial dysfunction, and cell death
[14][15][14,66]. With aging, the human retina undergoes various structural and physiologic changes
[16][67]. Aging has been associated with fewer retinal neurons (Rod PRs, RGCs, Rod bipolar cells), along with numerous age-related alterations, such as higher rate of mtDNA mutations and decreased density of cells and synapses
[17][18][68,69]. mtDNA becomes damaged with age and oxidative stress, and the absence of good repair mechanisms leads to accumulative mitochondrial dysfunction, which is recognized as a crucial pathogenic element in age-related ophthalmic diseases (
Figure 1), such as glaucoma, DR, and AMD
[19][70].
Figure 1. Role of mitochondrial dysfunction in the pathogenesis of glaucoma, diabetic retinopathy (DR), and age-related macular degeneration (AMD). Created with
Biorender.com.
2.1. Glaucoma
Progressive loss of vision is the outcome of glaucoma, a neurodegenerative disease of the optic nerve characterized by the accelerated death of RGCs and their axons. Since RGCs are located in the optic nerve, they are especially vulnerable to mitochondrial respiratory capacity damages
[20][21][71,72]. As the world’s population ages, experts predict that glaucoma’s prevalence will rise, ultimately reaching 111.8 million people by the year 2040
[22][73]. In common with other neurodegenerative disorders, increasing age is a major risk factor for the prevalence and incidence of glaucoma. The causes of RGC degeneration in glaucoma are probably multifactorial
[23][74]. The link between increased age and prevalence of glaucoma suggests that aging may make the optic nerve more susceptible to various stressors, which eventually results in RGC death and optic nerve degeneration
[24][75]. In the early stages of glaucoma development, oxidative stress contributes to cellular damage in the trabecular meshwork, changes in the homeostasis of nitric oxide and endothelin, and finally, a role in cellular death in the ganglion cells
[25][76]. An association between glaucoma and mitochondrial dysfunction has been suggested in a recent clinical study, where a 21% reduction in mitochondrial respiratory function and an increase in mtDNA mutations were observed in peripheral blood of patients with primary open-angle glaucoma
[26][27][77,78].
Mitochondrial dysfunction is present in glaucoma animal models prior to RGC death
[28][29][79,80], suggesting a primary effect for mitochondrial abnormalities in glaucoma onset and subsequent progressive loss of vision. In animals, elevated Intraocular Pressure (IOP), a primary feature of glaucoma, reduces antioxidant defenses and increases oxidative stress, which may predispose RGC to apoptosis and degeneration
[30][31][81,82]. Blocking ROS can reduce mitochondrial dysfunction, and thus, slow the progression of glaucoma. This is because ROS are byproducts of electron outflow along the electron transport chain during cell respiration
[32][33][83,84].
Exogenous application of ROS triggers RGC apoptosis in vitro via caspase-independent pathways
[34][85], while reduction of ROS generation temporarily protects RGCs from apoptosis
[35][86]. An important transcriptional factor in these events is Nrf2, which influences the expression of a diverse array of antioxidant pathways, such as glutathione, as well as cytoprotective genes. Another important regulator of the antioxidant defense system is PGC-1𝛼, which modulates the antioxidant proteins that are not regulated by Nrf2
[36][37][87,88]. PGC-1α has been shown to regulate astrocyte activities and RGC homeostasis
[38][89]. Moreover, SIRT1 overexpression in mice showed significantly higher RGC number compared to wild type
[39][90]. Preliminary research shows that resveratrol (a SIRT1 activator) treatment of mice at 250 mg/kg mitigates RGC loss and maintains pupillary light responses. SIRT1 controls vascular endothelial growth factor-A (VEGF-A) by activating hypoxia-inducible factor-2 alpha (HIF-2α), as was noticed in a study conducted on hypoxic choroidal endothelial cells by Balaiya et al. SIRT1 deacetylates (at p65 subunit) and reduces NF-kB signaling that protects neurons from amyloid beta induced toxicity in microglia
[39][40][41][90,91,92]. It has been suggested that as a potent SIRT1 activator, MOTS-c would have the same effect as Resveratrol on RGCs
[42][93]. Additionally, the MOTS-c peptide could induce antioxidant and cytoprotective genes expression by PGC-1𝛼 activation and NRF2 upregulation, which maybe potentially useful for glaucoma treatment. MOTS-c can target mitochondria adaptation to restore energy production in cells with impaired OXPHOS, and therefore, may provide a potential therapeutic approach in glaucoma
[43][94].
2.2. Diabetic Retinopathy
Photoreceptors, bipolar, horizontal, amacrine, and ganglion neurons, Müller cells, astrocytes, microglia, and pigment epithelial cells are all negatively impacted by diabetes
[44][45][95,96]. Different racial populations (mtDNA haplogroups) have different susceptibilities to develop diabetes
[46][47][97,98]. DR, the most prevalent microvascular complication of diabetes, occurs gradually within 15 years of diagnosis in as many as 50% of type I diabetics and 10% of the type II diabetic patients
[48][49][99,100]. However, hyperglycemia is the primary factor in the onset of DR in people with diabetes
[50][101]. ROS production, including superoxide and hydrogen peroxide, is increased in retinal tissues due to hyperglycemia.
Retinal neurodegeneration is an early event during the progression of DR. The pathogenesis of DR involves progressive dysfunction of retinal mitochondria in the setting of hyperglycemia, with mtDNA damage and accelerated apoptosis occurring in retinal capillary cells
[51][102]. In retinal tissues, hyperglycemia upregulates production of ROS, such as superoxide and hydrogen peroxide
[34][52][85,103]. The cascade of mitochondrial fragmentation, DNA damage, increased membrane potential heterogeneity, decreased oxygen consumption, and cytochrome c release is triggered by oxidative stress. By releasing cytochrome c, mitochondria initiate the caspase cascade, thereby committing the cell to apoptosis
[53][104]. Oxidative stress also triggers activation of NF-κB, which initiates the release of pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), IL-8, and IL-1β. Moreover, their expression levels are correlated with the severity of DR
[54][55][105,106].
Since the disease pathogenesis is partially attributed to mitochondrial dysfunction, treatment options should also consider restoring normal mitochondrial function by lowering oxidative stress
[56][107]. Thus, therapies that target multiple steps of oxidative stress and mitochondrial damage should provide a hope for the prevention of this multifactorial blinding complication of diabetes
[57][58][59][108,109,110]. The development of retinopathy, on the other hand, has been prevented in animal models of diabetes thanks to antioxidant supplementation and overexpression of mitochondrial antioxidant enzymes
[60][111]. The recognition of MOTS-c and its inverse relationship to age and hemoglobin A1c (HbA1c)
[4][61][4,112] provide further evidence for the importance of MDP in insulin sensitivity. Investigations by Ramanjaneya et al. showed that subjects with type II diabetes had significantly lower levels of circulating MOTS-c than controls
[62][113].
2.3. Age-Related Macular Degeneration
AMD is a developing neurodegenerative disease of the central macular region involving PRs, RPE, Bruch’s membrane (BrM), and the choroid
[63][64][114,115]. AMD affects approximately 20–30% of people over 75 years in the developed world and it is estimated to be increased to 288 million persons by 2040
[65][116]. Vascular inflammation and dysregulation, mitochondrial damage and accumulation of ROS, and RPE cell senescence are the molecular underpinnings of the transition from normal aging processes to pathological AMD
[66][117]. RPE dysfunction plays a crucial role in pathophysiology of AMD
[67][118] and oxidative stress is a key factor for triggering RPE degeneration
[68][119].
The high metabolic activity of RPE cells is due to their numerous mitochondria. By utilizing OXPHOS, mitochondria are the body’s primary source of energy production. Reduced energy production and increased apoptosis are both consequences of mitochondrial dysregulation, which is thought to be a root cause of AMD
[69][70][71][120,121,122]. These alterations lead to a decline in bioenergetics, an uptick in mitochondrial ROS production, mitochondrial dysfunction, and ultimately cell death. RPE cells isolated from AMD patients were found to be particularly vulnerable to mtDNA damage
[5][72][73][74][5,123,124,125].
Two central elements of RPE damage are NRF-2/ARE and PGC-1𝛼, which undergo downregulation and are associated with increased oxidative stress, lipofuscin accumulation, and mitochondrial damage involved in AMD pathophysiology
[75][76][126,127]. In patients with AMD, plasma levels of complement regulatory proteins Factor H and Factor I (encoded by the CFH and CFI genes, respectively) as well as some inflammatory cytokines (IL-6 and TNF-α) are elevated
[77][78][128,129].
There is solid evidence that dysfunction of the metabolic ecosystem leads to the retinal degeneration associated with AMD. In a large study on dysregulated metabolic pathways in AMD, Golestaneh et al. reported downregulated AMPK/SIRT1 and PGC-1α pathways along with overactive mTOR expression contributed to the underlying disease mechanisms in AMD RPE. These events could directly affect mitochondrial metabolism and biogenesis
[79][130]. Based on these studies, it is reasonable to speculate that pathways related to energy metabolism, mitochondrial biogenesis, and oxidative stress may be ideal targets to treat AMD. Activation of AMPK promotes downstream energy producing pathways, including glucose metabolism, mitochondrial function, and autophagy. This makes AMPK an ideal target for diseases such as AMD.
It has been suggested that mtDNA fragmentation and MDPs may play a role in the pathology of AMD, and this is supported by other studies. The mtDNA damage in AMD patients was found to be increased by 350% and localized to specific regions of the mitochondrial genome, including the 16S and 12S ribosomal RNA genes and eight of the mitochondrial genome’s 22 tRNA genes
[5]. Damage to 16S rRNA or 12S rRNA could result in dysregulated production of MDPs. HN and SHLPs, provide cytoprotective functions in RPE cells and ocular diseases and may be considered as potential therapeutic targets for AMD
[7][80][81][7,131,132]. MOTS-c is primarily expressed in the perinuclear region and the cytoplasm of RPE. In unstressed RPE cells, MOTS-c co-localized primarily with mitochondria, with negligible localization in the nucleus
[5][82][5,33]. Serum-starved RPE cells cotreated 24 h with tert-Butyl Hydroperoxide (tBH) (150 µM) and increasing doses of MOTS-c (1 µg to 10 µg) demonstrated elevated protection of RPE cells, with the highest dose providing the most protection.