Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jonathan Samuel Chávez-Iñiguez and Version 2 by Camila Xu.
Chronic kidney disease (CKD) is a highly prevalent condition worldwide in which the kidneys lose many abilities, such as the regulation of vitamin D (VD) metabolism. Moreover, people with CKD are at a higher risk of multifactorial VD deficiency, which has been extensively associated with poor outcomes, including bone disease, cardiovascular disease, and higher mortality.
- chronic kidney disease
- CKD-MBD
- calcitriol
- vitamin D
1. Introduction
Chronic kidney disease (CKD) is a highly prevalent condition worldwide in which the kidneys are functionally and/or structurally damaged [1][2][1,2]. As a result, the kidneys lose their ability to properly excrete waste products and perform certain specific endocrine functions [3]. For example, the kidneys are known to play a crucial role in regulating vitamin D (VD) metabolism by converting VD into its active form [1,25-dihydroxy-VD or calcitriol (CTR)] [3][4][3,4]. In people with CKD, this ability is impaired, not only because of the loss of functional kidney tissue as CKD progresses but also because of the important role of the multifactorial and early increase in fibroblast growth factor-23 (FGF23) [5][6][5,6]. FGF23 is a bone-derived hormone whose main target organ is the kidney, where it suppresses the transcription of the key activation enzyme, 1α-hydroxylase (CYP27B1), and activates the transcription of the key degradation enzyme, 24-hydroxylase (CYP24A1), in the proximal renal tubules [7][8][7,8], thereby leading to reduced availability of CTR. Moreover, the circulating concentration of CTR is a positive regulator of FGF23 secretion in bone, creating a feedback loop between kidney and bone. The intracellular signaling cascades downstream of the FGF receptors that regulate the transcription of these hydroxylases in the proximal renal tubules remain to be elucidated [7], as well as the effects of calcium on FGF23 metabolism [9]. It is also important to consider the early reduction of the important FGF23 cofactor klotho in CKD [10][11][10,11]. In the presence of klotho, the FGF23 protein gains bioactivity to influence phosphate (P) homeostasis and VD metabolism [12][13][12,13]. Among many other effects [13], independently of CKD itself [14], both increased levels of FGF23 and decreased levels of klotho have been clearly associated with mortality [15][16][17][15,16,17] and survival [18][19][18,19].
It is now known that VD deficiency [as defined by serum 25-hydroxy-VD (calcidiol) levels] is very common in the general population worldwide [20][21][20,21], and studies have shown that people with CKD are at a higher risk of multifactorial VD deficiency due to dietary restrictions and reduced sunlight exposure, among many other factors [22][23][22,23]. VD deficiency has been extensively associated with poor outcomes, including bone disease, cardiovascular disease, and higher mortality [24][25][26][24,25,26]. Indeed, a plethora of pleiotropic effects have been associated with VD, which is partly explained by the fact that extra-renal organs have the enzymatic capacity to convert calcidiol into CTR [22][27][22,27]. There is no doubt that the evidence is abundant in terms of the association of almost all negative outcomes with low levels of VD, above all the information poured from experimental studies [27][28][29][30][27,28,29,30]. In humans, retrospective cohorts, prospective studies, and even meta-analyses have found this association [28][29][30][31][28,29,30,31], but the recent VITAL randomized clinical trial (RCT) has significantly lowered the previous high expectations regarding the beneficial effects of VD supplementation in the general population [32], and RCTs in CKD have mostly failed—but not all—in their primary objectives [31][33][34][35][31,33,34,35].
2. CKD-MBD, Vitamin D, Skeletal Fragility, and Osteoporosis
Since all these complex VD pathophysiology pathways lead to important derangements in CKD, VD is still considered an integral part of the systemic CKD mineral and bone metabolism disorder, now known by the acronym CKD-MBD [36][37][38][39,43,44] (Figure 1). VD plays a vital role in maintaining bone health by promoting intestinal calcium absorption and regulating the activity of osteoblasts and osteoclasts [4][39][4,45]. In addition, VD is involved in many other genomic, biochemical, and clinical pathways, with stimulatory or inhibitory effects on the occurrence of morphological and/or functional changes in vital organs, which confer on VD its systemic functions beyond bone [39][40][41][45,46,47]. Specifically, VD deficiency is not merely one of the laboratory abnormalities in need of clinical monitoring but rather is associated with all of the other components (bone disease, vascular calcification, fractures, cardiovascular disease, and mortality) (Figure 1) [36][37][38][42][43][39,40,43,44,48]. Moreover, VD deficiency (native or active) has been associated with the development of the well-known CKD-associated secondary hyperparathyroidism (SHPT). In fact, 42%–80% of patients with CKD G3–G4 have SHPT with low serum calcidiol levels and/or other related pathophysiological factors (e.g., increasing P load) [4][36][42][44][45][46][47][48][49][4,39,40,49,50,51,52,53,54]. The incidence of SHPT increases with decreasing renal function, and VD deficiency is more common in patients with CKD than in the general population [36][42][44][50][39,40,49,55]. Both VD deficiency and SHPT have also been associated with an increased rate of CKD progression, cardiovascular events, and increased mortality [45][51][52][53][54][50,56,57,58,59]. This topic is becoming even more relevant with the increasing importance recently given to the diagnosis and potential treatment of skeletal fragility and osteoporosis in CKD patients [36][42][55][56][57][58][39,40,60,61,62,63] (Figure 1). In fact, a very important increase in the risk of fractures has been clearly recognized in patients with CKD, with a multifactorial predisposing factor of muscle weakness and/or risk of falling as contributory factors [36][42][55][56][59][60][61][39,40,60,61,64,65,66].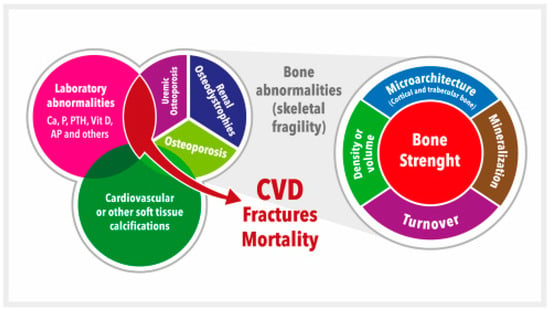
Figure 1. Schematic representation of the chronic kidney disease-mineral and bone disorder (CKD-MBD). CKD-MBD represents a systemic disorder of mineral and bone metabolism due to CKD manifested by either one or a combination of: (a) Laboratory abnormalities [calcium (Ca), phosphate (P), or vitamin D (vitD), among others (i.e., alkaline phosphatase, AP), etc.]; (b) Bone abnormalities in bone turnover, mineralization, volume, etc., ultimately affecting bone strength; and (c) Cardiovascular or other soft tissue calcifications. This figure illustrates the interrelated nature and consequences of CKD-MBD. The area occupied by different concepts is not associated with their relative importance. Adapted from S. Moe et al. (references [37][38][43,44]). CVD = Cardiovascular disease.
3. KDIGO Guidelines: From Vitamin D Deficiency to Osteoporosis Treatment
KDIGO (Kidney Disease: Initiative Global Outcomes) 2017 Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of CKD-MBD represented a selective update of the prior guideline published in 2009 [36][62][39,67]. Table 1 shows that, in the 2017 update, it was suggested that the potential presence of VD deficiency should be evaluated in patients with CKD G3a-G5 [glomerular filtration rate (GFR) ˂ 60 mL/min/1.73 m2, not on dialysis], among other modifiable factors, whenever intact parathyroid hormone (iPTH) levels are progressively rising or persistently above the upper normal limit (UNL) of the assay. VD deficiency is usually corrected with native VD (cholecalciferol, ergocalciferol, or even calcifediol), but dosage (daily, weekly, monthly) and targets are still matters of controversy, with variations among different guidelines [22][36][42][63][64][65][66][22,39,40,68,69,70,71]. For instance, in early 2011, a committee convened by the Institute of Medicine (IOM) issued a report on the Dietary Reference Intakes (DRI) for calcium and VD (Table 2) [64][65][69,70], and in July 2011, the Endocrine Society Task Force published a guideline for the evaluation, treatment, and prevention of VD deficiency [66][71]. Disagreements concerning the nature of the available data and the resulting conclusions led to some confusion [67][72], which may be even more evident if one also considers the presence of CKD [68][73].Table 1. Comparison of KDIGO guidelines 2009/2017 on treatment of abnormal PTH levels in non-dialysis patients. Adapted from reference [36][39].
| KDIGO 2009 | KDIGO 2017 | |
|---|---|---|
| Guideline 4.2.1 | In patients with CKD G3a-G5 not on dialysis, the optimal PTH level is not known. However, we suggest that patients with levels of intact PTH above the UNL of the assay be first evaluated for hyperphosphatemia, hypocalcemia, and vitamin D deficiency. (Evidence Level 2C) |
In patients with CKD G3a-G5 not on dialysis, the optimal PTH level is not known. However, we suggest that patients with levels of intact PTH progressively rising or persistently above the UNL for the assay be evaluated for modifiable factors, including hyperphosphatemia, hypocalcemia, high phosphate intake, and vitamin D deficiency. (Evidence Level 2C) |
| Guideline 4.2.2 | In patients with CKD G3a-G5 not on dialysis, in whom serum PTH is progressively rising and remains persistently above the UNL for the assay despite correction of modifiable factors, we suggest treatment with calcitriol or vitamin D analogs. (Evidence Level 2C) |
In adult patients with CKD G3a-G5 not on dialysis, we suggest that calcitriol and vitamin D analogs not be routinely used. (Evidence Level 2C). It is reasonable to reserve the use of calcitriol and vitamin D analogs for patients with CKD G4-G5 with severe and progressive hyperparathyroidism. (Not Graded) |
Table 2. Dietary Reference Intakes for Vitamin D (Institute of Medicine). Adapted from references [64][65][69,70]. The upper level intake for calcium has not been included since it is much greater than the upper limit considered safe in CKD patients (1000–1200 mg/day).
| Estimated Average Requirement (EAR) | Recommended Dietary Allowance (RDA) | Upper Level Intake | |
|---|---|---|---|
| 19–70 y/o | 400 IU/day The EAR for calcium varies from 800 mg/day (19–50 y/o females and 19–70 y/o males) to 1000 mg/day (51–70 y/o females) |
600 IU/day The RDA for calcium varies from 1000 mg/day (19–50 y/o females and 19–70 y/o males) to 1200 mg/day (51–70 y/o females) |
4000 IU/day |
| >70 y/o | 400 IU/day The EAR for calcium is 1000 mg/day |
800 IU/day The RDA for calcium is 1200 mg/day |
4000 IU/day |
y/o = years old; IU = international units.