The IL-10 family is composed of six members, namely IL-10, IL-19, IL-20, IL-22, IL-24, and IL-26. As the founding member, IL-10 has anti-inflammatory and immunosuppressive properties that serve to prevent excessive inflammation. It has been demonstrated to inhibit the antigen presentation capabilities of monocytes, macrophages, and dendritic cells, while simultaneously enhancing their tolerance-inducing, scavenger, and phagocytic functions. Additionally, IL-10 has been shown to suppress Th1-, Th2-, and Th17-mediated immune responses by inhibiting the proliferation of CD4+ T cells and their ability to produce proinflammatory cytokines. Furthermore, it has been observed to inhibit the secretion of proinflammatory mediators by neutrophils, eosinophils, and mast cells, as well as mast cell development.
2. Effects of Interleukins on Cardiomyocytes (CMs) in Heart Disease
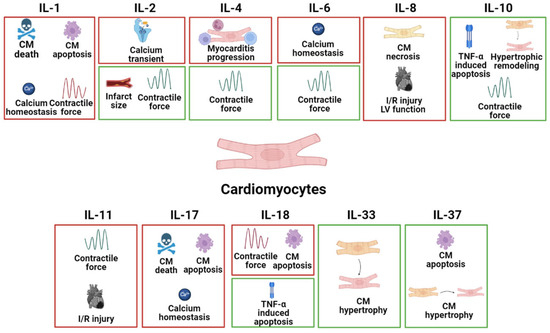
CMs are the beating muscle cells that make up the atria and ventricles and are being targeted primarily in heart disease therapy. The specific effects of different interleukins on CMs in common heart disease are listed in
Figure 1.
Figure 1. The specific pathophysiological effects of different interleukins on CMs in common heart disease. The box in red represents the deleterious role, and the box in green represents the protective role.
2.1. IL-1
IL-1α and IL-1β are proinflammatory cytokines and their levels are correlated with the severity and pathogenesis of heart disease. Targeting the IL-1 signaling cascade including IL-1α and IL-1β may be a promising therapeutic target for patients with MI
[12][13][12,13]. Moreover, in vivo MI mouse models have shown that inhibition of IL-1α reduces myocardial I/R damage, resulting in the retention of left ventricular function, reduced infarction area, and decreased activation of inflammatory bodies
[14]. Thus, IL-1α blockers may represent an effective therapeutic approach to reduce I/R damage to the heart.
In a mouse model of MI, dead cardiomyocytes will release IL-1α
[15][16][15,16]. In addition, the release of IL-1β in fulminant myocarditis leads to extensive inflammation, leading to the further death of cardiomyocytes, the gradual loss of active contracted tissue, and the development of cardiomyopathy and HF.
IL-1β is concentration-dependent and may elevate myocardial ring GMP through the myocardial L-arginine-NO pathway, leading to the restriction of systolic ejection and cardiac depression
[13]. The regulation of the excitatory-contractile coupling of cardiomyocytes is reflected in changes in contractile force, and cytokine-specific effects appear to exist in the excitatory–contraction coupling, with TNF-α and IL-1β affecting inward calcium currents
[17][18][17,18]. IL-1β has been shown to significantly prolong the duration of the action potential of guinea pig ventricular cells by changing the conductance of calcium channels
[19]. IL-1β has been shown to rapidly inhibit the voltage-dependent Ca
2+ current in adult rat ventricular muscle cells. Consistent with these data, IL-1β has been shown to inhibit systolic cardiomyocyte function by potentially involving the destruction of calcium processing or the inhibition of a β-adrenergic response
[20][21][20,21]. In patients with HF, IL-1β has been demonstrated to decrease the beta-adrenergic responsiveness of L-type calcium channels, as well as decrease calcium homeostasis genes, including phospholamban and sarcoplasmic reticulum calcium ATPase
[22][23][22,23]. Furthermore, IL-1β has been shown to have a proapoptotic effect on cardiomyocytes
[24] and exert negative inspiratory effects on both isolated cardiomyocytes and intact hearts
[13][25][13,25].
2.2. IL-2
Recent studies have demonstrated that high doses of IL-2 may induce AMI
[26]. In isolated normal myocytes, IL-2 was found to decrease the amplitude of calcium transients induced by electrical stimulation, likely through blocking Ca
2+ ATPase activity in the sarcoplasmic reticulum
[27]. Furthermore, IL-2 concentrations produced by CD4+ T lymphocytes were abnormally elevated in patients with DCM, which may reflect deficiencies in T-cell function in these patients
[28].
However, there are also studies demonstrating the protective role of IL-2 in MI. The injection of IL-2-activated NK cells has been shown to promote vascular remodeling through a4b7 integrin and killer cell lectin-like receptor (KLRG)-1 and promote cardiac repair after MI
[29][30][29,30]. In addition, Cao et al. have reported that IL-2 could reduce infarct size by activating kappa-opioid receptors
[31]. Moreover, IL-2 can be stimulated by the IL-2IgG2b fusion protein to improve left ventricular (LV) contraction function and remodeling in an MI rat model
[30].
In conclusion, additional experimental studies are needed to fully elucidate the role of IL-2 and develop its therapeutic potential in heart disease.
2.3. IL-4
IL-4 is generally regarded as an anti-inflammatory cytokine. A recent study by Wan et al. showed that Vγ1+ γδT cells, as one of the main early producers of IL-4 after acute viral infection, protect the mouse heart from acute viral myocarditis. Moreover, the neutralization of IL-4 in mice led to exacerbations of acute myocarditis, confirming the IL-4-mediated Vγ1 protective mechanism
[32]. This finding was further supported by another study on viral myocarditis, which showed elevated levels of IL-4 in mice with attenuated viral myocarditis and elevated levels of heart expression
[33].
However, a contradictory finding was reported in the context of autoimmune myocarditis, where eosinophils are the predominant cell type in the heart expressing IL-4, and eosinophil-specific IL-4 deletion leads to improved cardiac function. In this regard, eosinophils have been shown to drive myocarditis progression to inflammatory dilated cardiomyopathy (DCMi), and this process is mediated by IL-4
[34].
2.4. IL-6
IL-6, as an upstream marker of inflammation, is independently associated with the risk of major adverse cardiovascular disease events, MI, HF, and cancer mortality stable coronary heart disease
[35]. IL-6 and sIL-6R have been associated with AMI and cardiac injury; binding to trans-IL-6 receptors alters intracellular signaling, and blocking IL-6 receptor binding may be a causative factor in AMIs
[36]. Hypothetically validated, the IL-6 receptor antagonist tocilizumab reduces inflammation and the release of TnT in non-ST-segment elevation MI (NSTEMI). Therefore, IL-6 is a potential therapeutic target for MI
[37].
Cytokine-specific action appears to be present in the excitatory–contraction coupling and TNF-α IL-6 modulates Ca
2+ ATPase activity of the sarcoplasmic reticulum in cardiomyocytes
[38]. It was reported that the degradation of IL-6 mRNA inhibits the proinflammatory action in the stress-overloaded myocardium
[39]. Moreover, the gene deletion of IL-6 improves cardiac function and weakens hypertrophy by eliminating the dependent effects of CaMKII on cardiomyocytes in the stress-overloaded myocardium
[40]. In addition, in the model of LV remodeling after MI, a drug blockade of IL-6 through the administration of an anti-IL6R antibody weakens dilation and improves contraction function
[41]. IL-6 can indirectly enhance the expression of iNOS, and excess nitric oxide may reduce myocardial contractility and may have toxic effects by triggering apoptosis
[42].
IL-6 exerts negative inotropic action
[43] and promotes a hypertrophy response in cardiomyocytes
[44][45][46][44,45,46] through the gp130/STAT3 pathway, but can also enable protective action, mediated by mitochondrial function preservation
[47]. Recombinant IL-6 induces a cytoprotective effect to prevent I/R damage and activates ERK1/2, JNK1/2, p38-MAPK, and PI3K without inducing STAT1/3 phosphorylation. These data suggest that the cardiomyocyte protective effect of IL-6 in I/R occurs through ERK1/2 and PI3K activation, but is not related to sIL-6R and JAK/STAT signaling
[48].
2.5. IL-8
IL-8 is important in the development of MI. Serum IL-8 concentrations show a transient increase in the very early stages of AMI
[49]. Specific monoclonal antibodies that neutralize IL-8 significantly reduce the degree of necrosis in rabbit myocardial I/R injury models
[50]. High levels of IL-8 in STEMI patients with HF are associated with less improvement in left ventricular function in the first 6 weeks after PCI, suggesting that IL-8 may play a role in reperfusion-related injuries to the myocardium after ischemia
[51].
2.6. IL-10
Cardioprotective effects of IL-10 on the cardiomyocytes of heart diseases were found in a variety of previous studies. The involvement of the Akt and Jak/Stat pathways in regulating TNF-induced cardiomyocyte apoptosis by IL-10 has been studied
[52]. Subsequent research revealed that IL-10’s negative control of TNF-induced apoptosis was mediated by Akt via STAT3 activation
[53]. In addition, there is a study revealing that IL-10-induced antiapoptotic signaling in cardiomyocytes includes upregulating TLR4 through MyD88 activation
[54]. Moreover, Kishore, R et al. found that IL-10 attenuates pressure overload-induced hypertrophic remodeling and improves heart function via STAT3-dependent inhibition of NF-κB
[55]. Exercise reduces HFD-induced cardiomyopathy by reducing obesity, inducing IL-10, and reducing TNF-α
[56].
2.7. IL-11
IL-11 mediates cytoprotective signals in cardiomyocytes by activating phosphorylated STAT3 translocating into nuclei
[57]. IL-11 attenuated cardiac remodeling after MI through the gp130/STAT3 axis
[5]. In addition, it also reduced the I/R injury through STAT3 activation in the hearts
[58]. This evidence demonstrates the therapeutic role of IL-11 in heart disease.
2.8. IL-17
As a proinflammatory cytokine, IL-17 participates in an array of heart diseases. IL-17 was reported to have contributed to the process of cardiac fibrosis, the activation of matrix metalloproteinases, and enhanced cardiac cell death. IL-17 induces mouse cardiomyocyte apoptosis via Stat3-iNOS activation, suggesting that IL-17 contributes to cardiac damage
[59]. It has also been observed that IL-17A induces cardiomyocyte apoptosis through the p38 mitogen-activated protein kinase (MAPK)-p53-Bax signaling pathway and promotes both early- and late-phase post-MI ventricular remodeling
[60]. Pan et al. found that IL-17 affects the calcium-handling process involved in HF. The treatment of neonatal cardiomyocytes with steady-state concentrations of IL-17 suppressed transient calcium and decreased SERCA2a and Ca
v1.2 expression, this effect is mediated via the NF-κB pathway
[61].
2.9. IL-18
IL-18 is a proinflammatory cytokine produced during various heart diseases. IL-18 was discovered to be elevated in animal models of AMI, HF, pressure overload, and LPS-induced dysfunction. Furthermore, IL-18 has been shown to regulate cardiomyocyte hypertrophy, induce cardiac systolic dysfunction, and lead to extracellular matrix remodeling
[62][63][62,63]. Several observations suggest that IL-18BP is a potential therapeutic tool for reducing myocardial dysfunction caused by ischemia
[64][65][66][64,65,66].
The treatment of HL-1 cardiomyocytes with IL-18 resulted in hypertrophy and elevated levels of ANP, likely via the activation of signaling pathways involving PI3K, Akt, and the transcription factor GATA4
[67]. In vitro studies found that the IL-18 treatment of cardiomyocytes increased peak and diastolic calcium transients and decreased the shortening of isolated cardiomyocytes
[68]. Moreover, an increase in serum IL-18 concentration may induce apoptosis in cardiomyocytes, leading to ongoing myocardial injury in acute MI
[69].
However, there are some controversial studies; for example, the expression of IL-18RNA in the myocardium of patients with dilated cardiomyopathy is downregulated
[70], and IL-18 has been shown to play a beneficial role in viral myocarditis caused by the cerebrocarditis virus. The systemic administration of IL-18 is beneficial in mice with myocarditis and may be mediated by reducing the expression of TNF-α in the heart
[65].
Overall, the role of IL-18 in heart disease is primarily in amplifying myocardial dysfunction, and the level also was recognized as a marker of heart injury in patients.
2.10. IL-33
IL-33 belongs to the IL-1 family. IL-33 and its receptor ST2 (located on the membrane of CMs) were demonstrated to be cardioprotective. The highly localized signaling pathway mediated by ST2 regulates the heart’s response to pressure overload. It was suggested that IL-33 secretion by endothelial cells is crucial in converting myocardial pressure overload into a selective systemic inflammatory state
[71]. In a study involving wild-type mice, treatment with recombinant IL-33 was found to reduce angiotensin II and phenylephrine-induced cardiomyocyte hypertrophy and fibrosis. Furthermore, IL-33 treatment improved survival after transverse aortic constriction (TAC), a surgical procedure used to induce cardiac hypertrophy and heart failure in animal models
[72].
2.11. IL-37
IL-37, like IL-10, is an anti-inflammatory interleukin generated by a variety of cell types. IL-37 is the main cytokine in the regulation of immune response, mainly inhibits the expression, production, and effect of proinflammatory cytokines, and plays a role in autoimmune diseases and organ transplantation
[73]. The expression level of IL-37 is known to be low under normal physiological conditions; however, the expression level of IL-37 is significantly upregulated in response to an inflammatory environment, such as in patients with acute myocardial infarction (AMI)
[74].
IL-37 plays an active role in a variety of cardiovascular diseases
[75]. The Zeng group reported that human recombinant IL-37 can inhibit neutrophil infiltration and reduce cardiomyocyte apoptosis through a tail vein injection into myocardial I/R mice, thereby alleviating myocardial I/R injury in mice
[76]. The team also reported that the intraperitoneal injection of human recombinant IL-37 and the intravenous injection of IL-37 and troponin co-induced dendritic cells can alleviate adverse ventricular remodeling after MI and cardiomyocyte apoptosis in mice, also attenuating the degree of cardiac fibrosis
[77]. Overall, the positive role of IL-37 on other heart diseases, i.e., HF, needs to be further elucidated.
Overall, the effects of interleukins on CMs in heart disease are complex and context-dependent, and more research is needed to fully understand their roles in these conditions.