1. Introduction
The epigenome encompasses multiple interacting regulatory elements that define phenotypic variation beyond what the DNA sequence encodes
[1][2][3][1,2,3]. In addition to their roles in development and differentiation, epigenetic mechanisms play a fundamental role in transcriptional regulation during disease progression, further underscoring the importance of understanding their molecular bases
[4][5][4,5]. Chromatin is a complex of proteins and nucleic acids found in the eukaryotic cell nucleus and is subjected to continuous changes to accommodate cellular metabolic needs. As chromatin is the physiological template of the majority of the epigenetic mechanisms, it refers to heritable and reversible changes that mediate all DNA-dependent processes, including replication, repair, recombination and transcription, without altering the DNA sequence. In a versatile and highly coordinated manner, epigenetic regulation of transcription establishes cell-specific gene expression signatures from the same genome. It allows the cell to change these gene expression signatures in response to stimuli, such as changing conditions from their environment
[2]. Some of the best-characterized epigenetic mechanisms of transcriptional regulation involve DNA methylation, histone modifications, nucleosome remodeling, regulation via noncoding RNAs (ncRNAs) and nuclear matrix interactions
[5][6][7][8][5,6,7,8].
2. Epigenetics in the Context of Environmental Exposure
DNA methylation in eukaryotes is mediated by DNA methyltransferases (DNMTs) and refers to the covalent transfer of a methyl group to the C5 position of cytosine forming 5-methylcytosine (5mC), most frequently at the dinucleotide sequence CG (mCG)
[9]. DNA regions that are ≥200 bp long and show a CG:GC ratio ≥0.6 are defined as CpG (citosine-phosphate-guanosine) islands (CGIs), which are often located within the promoter of protein-coding genes
[10]. It is noted that methylation reprogramming can result from the inhibition of DNMTs or de novo DNMTs activity. This mechanism is reversible and can be mediated by different mechanisms involving DNA repair and ten-eleven translocation (TET) enzymes, which catalyze the oxidation of 5mC, forming several intermediates such as 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) until its full conversion to cytosine
[11][12][11,12]. Although 5hmC, 5fC and 5caC are less abundant than 5mC, they contribute to a sensitive and dynamic read-out of cell state as their profiles are in part determined by an active gene–body transcription and enhancer activity, which are rapidly altered upon environmental challenge
[13].
The genomic landscape of 5mC is the product of the constant activity of both DNA methylation and demethylation processes, resulting in a dynamic equilibrium that can be shifted in response to stimuli, including changes in the external environment of the cell. While the loss of 5mC is associated with genomic instability and cancer, the gain of 5mC has been associated with several congenital defects and other diseases, thus highlighting not only the importance of the balance of DNA methylation but also the possible consequences of their alteration
[14][15][16][14,15,16]. Changes in nutritional status and environmental exposure to several agents can modify genomic DNA methylation patterns, thereby affecting chromatin structure and gene expression toward disease
[15][17][15,17]. Patterns of 5hmC directly correlate with gene expression profiles and can be rapidly altered following short-term exposure to environmental agents, in a proportional manner to the duration of the exposure. For instance, several 5hmC changes induced by the nongenotoxic carcinogen phenobarbital (PB) were shown to persist in PB-driven tumors; thus, such changes represent early exposure biomarkers that are maintained across cellular progenies
[13]. Interestingly, loci enriched with 5hmC after exposure to PB correspond mostly to enhancers and in less quantity to promoters and gene bodies. Furthermore, PB-induced aberrant levels of 5hmC have been associated with liver tumors in mice, therefore suggesting that PB exerts its pathological effect through epigenetic regulation, leading to disease
[18][19][18,19].
Posttranslational modifications of histone proteins (further referred to as histone modifications) constitute another level of epigenetic mechanisms of transcriptional regulation. Histone proteins (H1, H2A, H2B, H3 and H4) are relatively small and basic proteins that are abundant in the cell nucleus and are an essential part of the nucleosome. The nucleosome is the basic repeating structural and functional unit of chromatin, consisting of the nucleosome core particle, the linker DNA between two nucleosome core particles and an H1 molecule
[20]. The nucleosome core particle consists of approximately 146 base pairs of genomic DNA wrapped around a histone octamer of two H2A-H2B dimers and one (H3-H4)2 tetramer
[21]. Due to the structural features of the nucleosome, histone proteins can undergo posttranslational modifications at their N-terminal tails, which comprise acetylation, methylation, phosphorylation, ubiquitination and sumoylation, among others
[22][23][24][25][22,23,24,25]. While DNA methylation is relatively stable in somatic cells, histone modifications are more diverse and dynamic, changing rapidly during the cell cycle
[6][8][22][23][6,8,22,23]. Acetylation at specific amino acids of histones (e.g., histone 3 lysine 9 acetylation, H3K9Ac) is generally associated with active chromatin. It is mediated by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs). Histone methylation also occurs at specific amino acids of histone proteins and can be associated with both repression (e.g., H3 lysine 27 trimethylation, H3K27me3) or activation (e.g., H3 lysine 4 trimethylation, H3K4me3) of gene expression. Various enzymes mediate histone methylation (histone methyltransferases; HMTs) and histone demethylation, represented mainly by histone lysine demethylases (KDMs)
[26][27][26,27].
Several environmental agents induce changes in histone modifications, thereby leading to changes in gene expression signatures. It has been reported that tobacco smoke influences histone methylation patterns. Further, the cadmium contained in tobacco smoke has been reported to have an impact on epigenetic landscapes in fetuses from smoking mothers. Similarly, exposure to lead (Pb) has been reported to have an impact on neurological disease via alterations in epigenetic regulation
[28][29][30][28,29,30]. On the other side, epigenetic modifications require metabolites as substrates. S-adenosylmethionine (SAM) is a common donor of methyl groups. The sources of SAM can be obtained from several metabolic routes such as glycolysis, amino acid metabolism, as well as folate and choline. It has been observed that a reduced methionine intake is associated with a decrease in methylation levels. In addition, the demethylation process requires α-ketoglutarate as a substrate. Furthermore, acetylation processes require acetyl-CoA as a substrate. When there is a high availability of acetyl-CoA, an increase in histone acetylation levels is also observed. The evidence strongly suggests that epigenetic regulation is closely related to nutritional factors
[31][32][33][31,32,33]. Perhaps one of the best-known examples of the effects of nutrition on epigenetics and health is the Dutch famine. In a harsh winter during World War II, caloric intake was severely reduced from what is recommended as healthy for human beings. Fetuses gestated under these circumstances showed as adults a higher incidence of obesity, type 2 diabetes, cardiovascular diseases, a propensity to dyslipidemias and even mental disorders. Remarkably, individuals exposed to these conditions showed differentially methylated loci in their genome, known as malnutrition-associated differentially methylated regions (P-DMRs). Such P-DMRs are frequently located in regulatory elements and particularly in regions associated with birth weight and LDL-cholesterol levels
[29][34][29,34].
Another component of epigenetic mechanisms that plays a significant role in mediating transcription involves ncRNAs. The majority of RNAs transcribed from the mammalian genomes are ncRNAs, which are not translated into proteins
[35]. ncRNAs can be classified based on their nucleotide length into small noncoding RNAs (sncRNAs, 21–34 nucleotides long) and long noncoding RNAs (lncRNAs, >200 nucleotides long)
[36][37][36,37]. Further classification of the ncRNAs can be done based on the biological processes in which they are involved, leading to a variety of ncRNA subtypes, including short interfering RNAs (siRNAs), transfer RNAs (tRNAs), Y RNAs, PIWI-interacting RNAs (piRNAs) and microRNAs (miRNAs) in the fractions spanning 15–40 nucleotides, while ribosomal RNAs (rRNAs) and lncRNAs are predominant in the fractions spanning ≥40 nucleotides
[38]. Epigenetic regulation of gene expression signatures by ncRNAs has been mainly related to lncRNAs and miRNAs. miRNAs constitute an RNA subtype of 21 to 25 nucleotides in length that act primarily in the cytosol by inhibiting translation
[39]. However, accumulating reports in the last decade demonstrate the presence of functional miRNAs in the cell nucleus as regulators of various biological processes, including transcription
[7][40][41][42][43][44][7,40,41,42,43,44]. For lncRNAs, different mechanisms have been characterized in detail for their roles in the nucleus
[35][45][46][47][48][35,45,46,47,48]. Through their interaction with specific genomic regions and proteins, they tether their regulated genes to specialized regions of the nucleus in which specific biological processes take place. For example, ncRNAs provide a framework for the assembly of defined chromatin structures at specific loci, thereby modulating gene expression, centromere function and the silencing of repetitive DNA elements. Exposure to diverse environmental factors also influences the epigenetic mechanism of the regulation of gene expression mediated by ncRNAs. Recent evidence suggests that heavy metals carry out their toxic action through miRNAs, particularly related to altered epigenetic mechanisms of gene expression in neurological diseases. Lead (Pb) and cadmium (Cd) are good examples of heavy metals that have been associated with the development of Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS)
[28][30][49][28,30,49]. From a nutritional perspective, it has been described that glucose intake activates thioredoxin-interacting protein (TXNIP), which induces
miR-204 expression. In turn,
miR-204 targets MAFA, a key transcription factor for insulin production, contributing to type 2 diabetes mellitus development
[50].
3. Relationship between Epigenetics and Oxidative Stress in the Context of Environmental Exposure
Exposure to different environmental factors, including pollutants, can change the epigenome and lead to adverse health effects. Pollutants such as heavy metals, endocrine disruptors (EDC), particulate matter (PM) and titanium oxide (TNM), among others, have been linked to epigenetic changes, including DNA methylation, histone modifications and ncRNA aberrant expression
[51][52][51,52]. However, oxidative stress (OS) is arguably the most common mechanism in the toxicology of environmental agents, unifying the action of broad classes of disparate environmental physicochemical pollutants, including oxidant gases, organic compounds, particulate surfaces and metal ions
[53].
OS results from environmental disturbance, which is known to modify cellular processes such as apoptosis, signal transduction cascades and DNA repair mechanisms
[54]. As OS is a natural physiological process where the presence of oxidants (reactive oxygen-derived species, ROS and nitrogen-derived species, RNS) overcome the uptake strategy of antioxidant defenses, it plays an essential role in epigenetic changes induced by environmental pollutants and culminates in signaling the disruption of redox control (
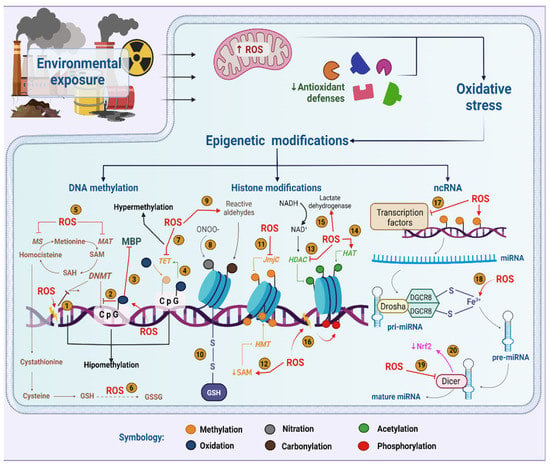
Figure 1)
[55][56][55,56].
Figure 1. Role of oxidative stress in epigenetic modifications induced by environmental pollutants. ROS potentially interfere with the activity of DNMTs (1), their ability to methylate cytokines (2) or their union with the MBP complex (3). Guanine oxidation makes the methylated cytokine more susceptible to oxidation by TET enzymes (4). In addition, ROS deplete SAM by oxidizing MAT and MS (5) or by using homocysteine to regenerate GSH (6) causing DNA hypomethylation. However, ROS can also inhibit TETs (7). ONOO- nitrates histones (8), while reactive aldehydes modify histones by carbonylation (9). Glutathionylation is a relevant modification in histones due to oxidative stress (10). In addition, ROS either inhibit JmjC demethylases (11) or attenuate HMTs activity by decreased SAM (12). Additionally, ROS inhibit HDACs (13) and stimulate HATs (14). However, ROS could activate HDACs through an increase in NAD+ (15). Histones can be phosphorylated upon DNA damage (16). ROS causes deregulation of transcription factors (17), pre-miRNA synthesis by interaction with Fe3+ (18) and Dicer activity inhibition, which impairs miRNA maturation (19) and NRF2 levels (20). Created with biorender.com.
It is noted that mitochondria are the primary intracellular source of ROS generation due to electron transfer during adenosine triphosphate (ATP) production
[57][58][59][57,58,59]. Complexes I and III of the electron transport chain (ETC) have been described as important sites of ROS production
[60][61][62][60,61,62]. These sites within ETC can leak even under normal conditions. The electron leak from the ETC reduces molecular oxygen (O
2) to superoxide anion (O
2−), which triggers the production of hydrogen peroxide (H
2O
2), which, in turn, can receive another electron and form the hydroxyl radical (-OH)
[63]. On the other hand, O
2− can also react with nitric oxide (NO), generating peroxynitrite anion (ONOO
−)
[64]. ONOO
− and OH are highly reactive ROS that induce oxidative damage to proteins, lipids and DNA when their production is exceeded
[65]. Therefore, to reduce the cellular damage caused by ROS, a cellular redox balance is needed. This balance is achieved by different antioxidants, including enzymes such as superoxide dismutase (SOD), glutathione peroxidase (GPx), catalase (CAT) and nonenzymatic antioxidants such as glutathione (GSH) and vitamins A, C and E
[66]. The expression of antioxidant enzymes and those associated with GSH production depends on different transcription factors, such as nuclear-erythroid-factor-related factor 2 (NRF2) and hairpin box O (FOXO). They both respond rapidly to oxidative environments to induce a redox balance, such as the expression of the abovementioned antioxidant enzymes
[66].
4. Microenvironment and Nutritional Influence on the Preservation of Epigenetic Marks
When cells are cultured
in vitro, they live and grow in conditions very different from the physiologic environment. Therefore, facing the need to adapt, cells use metabolic routes that allow them to survive and even thrive in the new media. Such adaptations result from epigenetic events
[67][168]. Several studies have demonstrated that cells that proliferate in culture tend to show changes in their epigenetic landscape
[68][69][70][71][72][169,170,171,172,173]. These changes are not random but, in fact, positively selected for the cell to adapt to the new media and thrive in it
[70][171].
5. Effects of Dietary Nutrients on the Epigenome
Dietary vitamins can modulate the epigenome and therefore have a direct impact on physiological and pathological processes
[73][74][255,256].
Vitamin C is an essential micronutrient that blocks oncogenic transformation induced by carcinogens
[75][257]. The protective role of vitamin C in cancer progression has historically been attributed to its antioxidant activity and the prevention of DNA damage induced by oxidative stress
[76][258]. Vitamin C is suggested to affect the genome activity via regulating epigenetic processes. For example, it has been identified that Fe and 2OG-dependent dioxygenases that catalyze the hydroxylation of methylated DNA and RNA and histones require ascorbate as a cofactor to initiate demethylation
[77][78][259,260]. It is a cofactor for TET dioxygenases that catalyze the oxidation of 5mC into 5hmC. Ascorbate is also required for the JmjC-domain-containing histone demethylases, serving as a cofactor for histone demethylation. In this manner, vitamin C appears to be a mediator between the genome and the environment. These findings demonstrate an unknown function of vitamin C in regulating the epigenome, which needs a re-evaluation of the functions of vitamin C in human health and disease
[75][257]. Recent studies have shown that vitamin C, by enhancing TET activity, can directly influence DNA methylation levels that alter chromatin structure and the expression of tumor suppressors and DNA repair enzymes
[79][261].
Vitamin A (retinol) and its metabolite all-trans retinoic acid reduce DNA methylation by raising the level of TET proteins (which oxidize DNA methylation) and, in doing so, promote stemness
[80][262]. In embryonic stem cells, Vitamin A can displace HDACs from binding to retinoic acid response elements within the promoter of target genes, thus promoting their expression
[81][263]. In neuroblastoma cells, Vitamin-A-induced transcription of the proto-oncogene
RET is associated with chromatin remodeling and demethylation of H3K27me3 of the enhancer, as well as increased H3K4me3 at the promoter region
[82][264].
Vitamin B12 is an essential cofactor for the synthesis of methionine (from homocysteine), which is critical for DNA methylation, yet methionine supplement cannot rescue the epigenetic hypomethylation of CpG associated with vitamin B12 deficiency
[83][265]. Deficiency of Vitamin B12 during pregnancy has epigenetic impacts on the next generation, leading to decreased global DNA methylation and increased expression of certain microRNAs such as
miR-221 and
miR-133 [84][266].
Vitamin D is a critical nutrient essential for human health and its deficiency is nowadays a major health problem. It is introduced with food or as a supplement, yet it is largely synthesized in the body upon exposure to solar ultraviolet B radiation. It has been calculated that the vitamin D axis can regulate up to 3% of the genome
[85][267]. The primary epigenetic effects of vitamin D are linked to histone acetylation
[86][268] and DNA methylation
[87][269]. In addition, vitamin D functioning through its receptor VDR is regulated by ncRNAs and, most importantly, VDR itself influences the expression of oncogenic and tumor suppressor lncRNAs
[88][270]. Under the category of
Vitamin E, saturated tocopherols and unsaturated tocotrienols with ROS scavenging properties are included. α-Tocopherol was reported to increase the methylation of the
miR-9 promoter, a miRNA involved in the control of glycemia
[89][271].
Dietary phytochemicals have antioxidant and anti-inflammatory properties and their prolonged consumption can leave long-lasting epigenetic marks on DNA
[90][91][272,273] and changes in ncRNAs levels
[92][93][274,275]. Particularly curcumin, quercetin, resveratrol and EGCG (EpiGalloCathechinGallate), among others, are well-known anti-inflammatory and antioxidant polyphenols and are formidable epigenetic modulators capable of regulating the activity of DNMTs, HATs and HDACs and also the biogenesis of microRNAs
[90][92][272,274]. Such epigenetic activities have been demonstrated on the basis of the curative potential of these dietary polyphenols in inflammatory diseases
[94][276], cardiovascular diseases
[95][277], metabolic disorders
[96][278] and cancer
[97][98][99][100][253,279,280,281].