Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Laura Cerchia and Version 2 by Dean Liu.
Aptamer-based immunotherapy has great potential to overcome significant challenges in T cell immunotherapy for solid tumors mainly represented by strong immunosuppressive signals, which induce low T cell activation and decreased synthesis and release of cytotoxic proteins.
- immune system
- immunotherapy
- aptamer
- TNBC
1. Tumor-Infiltrating Lymphocytes
The major types of immune cells in the TNBC microenvironment are TILs, and their presence is significantly associated with better survival outcomes in patients with early-stage untreated tumors [1]. TILs include all CD3+ T cells, which may promote tumor destruction (CD8+ cytotoxic T cells) and an antitumor response (CD4+ T-helper 1) or limit antitumor immune responses (CD4+ T-helper 2, including Forkhead box P3 (FOXP3) CD4+ regulatory T cells) [2][3].
Recently, Zhao et al. proposed an original strategy that exploits the targeting capability of aptamers to construct a “super-cytotoxic T lymphocyte” for enhanced antitumor response in cancer immunotherapy [4]. They generated acid-degradable metal–organic-based and lysosome-targeting nanoparticles that were loaded with perforin and granzyme B, two antitumor toxins contained in lysosomes of CD8+ T cells, and functionalized with an aptamer targeting the CD63 receptor on lysosome. Ca2+ was deposited on the nanoplatform to improve its biocompatibility and stability and potentiate toxin activity. The authors succeeded in using such an aptamer-guided platform (named LYS-NPs) for enriching lysosomes’ cytotoxic content of CD8+ T cells. When tested in the TNBC 4T1 mouse model, T cells preactivated with processed 4T1-specific antigens and recombined by LYS-NPs and released the lysosomal content into immunological synapses, triggering a strong antitumor reaction. The proposed aptamer-based immunotherapy has great potential to overcome significant challenges in T cell immunotherapy for solid tumors mainly represented by strong immunosuppressive signals, which induce low T cell activation and decreased synthesis and release of cytotoxic proteins [5].
2. Immune Checkpoint-Expressing Cells
Alatrash’s group reported that the expression of the PD-L1 gene in TNBC patients is significantly higher than in non-TNBC [6]. PD-L1, one of the major tumor cell-associated immune checkpoints, is expressed in a variety of immune cells, such as macrophages, some activated T cells, B cells, and in many solid tumor cells, including BC cells. Its receptor, the transmembrane protein PD-1, is expressed on the membrane surface of TILs, NK cells, macrophages, dendritic cells, and monocytes [7]. Binding between PD-L1 and PD-1 causes the inhibition of CD8+ TILs, transforming them into an anergic form and, consequently, cancer immune evasion.
Moreover, the PD-1/PD-L1 axis modulates within tumor cells various proliferative and survival signaling pathways such as PI3K/AKT, MAPK, JAK/STAT [8], and, very importantly, in TNBC, the activation of this axis promotes epithelial–mesenchymal transition (EMT), a phenotype associated with highly aggressive and metastatic tumors [9].
Different aptamer-based approaches in TNBC are currently being explored to revert PD-1/PD-L1 effects (Figure 1).
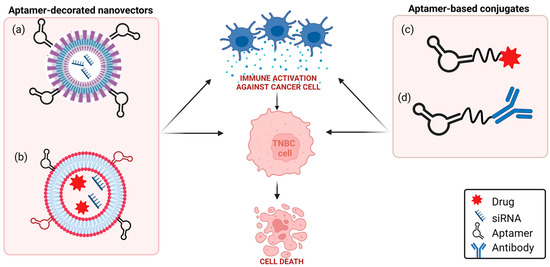
Figure 1. Schematic representation of aptamer-based strategies to block PD-1/PD-L1 axis in TNBC. (a) TNBC aptamer-decorated nanoparticles loaded with anti-PD-L1 siRNA; (b) anti-CD44 and anti-PD-L1 aptamer-decorated liposomes loaded with both doxorubicin and anti-IDO1 siRNA; (c) anti-PD-L1 aptamer conjugated to paclitaxel; (d) anti-EGFR aptamer covalently linked to anti-PD-L1 or anti-CTLA-4 mAbs (see text for details). Created with BioRender.com (accessed on 2 March 2023).
In this context, theour group investigated, for the first time, a combination between an anti-PD-L1 mAb with an anti-platelet-derived growth factor receptor β (PDGFRβ) aptamer, named Gint4.T, in TNBC [10]. Gint4.T is a nuclease-resistant 2′-fluoropyrimidines (2′F-Py) RNA aptamer which binds to and inhibits PDGFRβ expressed on the surface of different human cancer cells, including TNBC cells [11], and TNBC TME components, including mesenchymal stem cells [12], and T cells [10]. Interestingly, when intravenously injected in TNBC 4T1 syngeneic mice, the aptamer strongly potentiates the effect of anti-PD-L1 mAbs in inhibiting tumor growth and lung metastases formation by acting on both tumor cells and TME components [10]. Furthermore, the combined blockade of PDGFRβ and PD-L1 causes the depletion of FOXP3+ Treg cells and an increase in CD8+ T cells and granzyme B more consistently than single monotherapies. These results lay the foundation to construct a bispecific immunoconjugate consisting of an anti-PD-L1 antibody covalently linked to Gint4.T aptamer, thus optimizing the effectiveness of combination therapy. Bispecific constructs obtained by covalently linking an anti-epidermal growth factor receptor (EGFR) 2′F-Py RNA aptamer to immunomodulators anti-PD-L1 (10_12) [13] or anti-CTLA-4 (ipilimumab) [14] mAbs were generated by Passariello et al. and proved to maintain the biological functions of both parental moieties, thus exerting a potent cytotoxic activity against BC cells.
An alternative strategy to anti-PD-L1 mAbs for PD-L1 targeting is represented by the suppression of PD-L1 through gene silencing, which has the potential to overcome some recurring obstacles of mAbs-based treatments, such as their time- and cost-consuming production, the potential for immunogenicity, and low stability. Furthermore, this strategy allows to block the intrinsic pro-tumorigenic role of cytoplasmic PD-L1 [15] that, instead, is not accessible by antibodies. The possibility of synthesizing cancer-cell-targeting aptamers with functional groups at their extremity, allowing the conjugation to nanovectors, is a striking approach to deliver, specifically to the tumor, small-interfering RNA (siRNA) cargos, loaded in the nanovector, thus overcoming the vulnerability of siRNAs to nucleases and their inability to enter into target cells. Recently, poly(lactic-co-glycolic)-block-PEG (PLGA-b-PEG)-based nanoparticles have been loaded with anti-PD-L1 siRNA and decorated with a 2′F-Py RNA aptamer able to bind and internalize specifically into TNBC cells [16][17]. The resulting aptamer-conjugated nanovectors, upon 90 min incubation on TNBC cells, efficiently delivered siRNA into target cells, which was competent to cause an almost complete suppression of PD-L1 expression [18]. Notably, aptamer-decorated nanocarriers offer the possibility to link different ligands to the surface of the NPs, thus increasing the specificity of targeting, and to encapsulate in the NPs multiple therapeutics, thus allowing for efficacious combined therapies. For example, the concomitant administration of cisplatin [19] and siPD-L1 [18] by the PLGA polymeric nanoparticles, which rwesearchers equipped with TNBC aptamers, may not only promote a reduction in toxic side effects but also counteract the reported negative effect of cisplatin administration on the enrichment of PD-L1+ immune evasive TNBC cells [20]. In this regard, Kim et al. prepared a multifunctional nanosystem having two DNA aptamers conjugated on the external surface of liposomes and two different therapeutics inside nanovectors for synergistic chemoimmunotherapy in TNBC [21]. Specifically, they used, for TNBC cells targeting, the previously selected anti-CD44 [22] and anti-PD-L1 [23] DNA aptamers, each thiol-modified and covalently conjugated to maleimide groups of PEGylated-DSPE micelles by thiol–maleimide chemistry. Nanosized liposomes were loaded with both doxorubicin and siRNA interfering with the expression of IDO1, a protein that favors an immunosuppressive TME and is upregulated by doxorubicin treatment. When intravenously injected into TNBC 4T1 tumor-xenograft mice, the nanovectors strongly reduced tumor growth and inhibited metastasis formation by synergistically combining cancer-cell-targeted immunogenic cell death induction and reversal of immunosuppression [21].
Recently, different PD-L1 aptamers have been generated and tested as stand-alone antagonists, bispecific conjugates, and delivery agents of therapeutics in lung, liver, and colon tumor mouse models, which, similar to anti-PD-L1 antibodies, interfere with the PD-1/PD-L1 axis by blocking the PD-L1 (Table 1). One aptamer, named XQ-P3, has been generated by positive selection on PD-L1 overexpressing MDA-MB-231 cells by using PD-L1 knockout cells for counterselection [24]. Even if not yet tested in vivo, it appears highly effective in co-cultures of TNBC MDA-MB-231 cells and immune Jurkat cells by blocking the interaction with PD-1 and restoring T cell function. Furthermore, an XP-Q3 aptamer-paclitaxel conjugate showed anti-proliferation efficacy in PD-L1 overexpressed TNBC cells [24].
Table 1. Summary of anti-PD-L1 aptamers as cancer therapeutics.
| Aptamer | SELEX Approach |
Target (Positive Selection) |
Composition | Application | Ref. |
|---|---|---|---|---|---|
| AptPD-L1 | Nitrocellulose filter SELEX |
Recombinant human PD-L1 | DNA |
|
[23] |
|
[25] | ||||
|
[26] | ||||
|
[27] | ||||
|
[21] | ||||
|
[28][29] | ||||
| XQ-P3 | Cell-SELEX | PD-L1 overexpressing MDA-MB-231 cells | DNA |
|
[24] |
| N5, S42 | Capillary electrophoresis SELEX |
Recombinant human PD-L1 | Threose nucleic acid |
|
[30] |
| PL1 | Cell-SELEX | PD-L1 overexpressing CHO-K1 cells | Phosphorothioate DNA |
|
[31] |
Some aptamers against PD-1 receptor have also been selected and tested in mouse models of human cancers but not yet in TNBC (reviewed in [32]).
3. Macrophages
Tumor-associated macrophages (TAMs) are among the most abundant immune cells in the TME of a broad range of cancers and may act to promote or suppress antitumor immune responses [33][34]. Indeed, due to their high degree of plasticity, they shift to two diverse phenotypes in response to various micro-environmental stimuli: classically activated, pro-inflammatory M1, and alternatively activated anti-inflammatory M2, which display a differential expression profile to cell surface markers and different cytokine and chemokine production. M1 macrophages typically exert antitumor functions, while M2 macrophages promote tumor progression. In most aggressive tumors, including TNBC, TAMs tend to resemble an M2-like phenotype that largely accounts for the failure of conventional therapies and immune checkpoint inhibition therapies. For this reason, several innovative immunotherapeutic approaches aim to target and deplete M2 macrophages or reprogram them to the desired phenotype [35][36].
In order to select aptamers targeting human M2-like macrophages, the first cell-SELEX approach was applied to human macrophages derived from monocytes of several donors and polarized to the M2-like phenotype [37]. Although the best M2-targeting DNA aptamer coming from the selection was not able to discriminate the target cells from undifferentiated M0-like and monocytes and also bound at a lower extent to M1-like macrophages, it rapidly internalized into CD14+ monocytes, thus holding potential for monocyte-targeted drug delivery applications.
Another striking application of aptamers for solid tumor immunotherapy consists of potentiating M1 macrophage specificity for tumor cells by engineering them with cancer-cell-targeting aptamers. Chimeric antigen receptor T (CAR-T) cell immunotherapy, which infuses patients with CAR-T cells, has shown great efficacy in the treatment of some leukemias and lymphomas but only modest results in solid tumors due to the difficulty of penetrating tumors [38]. Because of the intrinsic capacity of macrophages to penetrate tumor tissues, several approaches have been recently proposed that genetically engineer them to express chimeric CARs (CAR-M) for targeting tumor cells and initiate a targeted antitumor response [39]. In order to overcome major drawbacks associated with traditional CAR-M therapies, such as the low reproducibility of engineered proteins and safety issues, Qian et al. proposed a new CAR-M approach based on the use of aptamers [40]. The murine stable macrophage cell line, RAW 264.7, was first incubated with an azide-containing metabolic glycoprotein labeling reagent and lipopolysaccharide to generate azido sugars on the M1 cell surface. Then, M1 cells were conjugated by click chemistry reaction to both the AS1411 aptamer, which binds to nucleolin expressed on several cancer cells, and a PD-L1 aptamer, for simultaneous tumor targeting and immune checkpoint blockade. Importantly, in vivo imaging of mice bearing 4T1 TNBC and intravenously injected with M1 cells, functionalized with fluorescent aptamers, showed a greater accumulation in tumors compared with unmodified M1 cells. Furthermore, when tested for antitumor activity, the dual-aptamer-engineered M1 caused a strong reduction in tumor growth and metastasis formation, which was accompanied by immune TME reprogramming with increased T cell infiltration in the tumor and enhanced T cell cytotoxicity.
Alternatively, Chen et al. proposed polyvalent spherical aptamers (PSAs) as a macrophage engineering strategy [41]. PSAs were generated through the functionalization of gold nanoparticles with both the thiol-modified AS1411 aptamer and a DNA linker that carries, at the free extremity, a functional group for reacting with azide tags created on M0 macrophages through the abovementioned metabolic labeling and biorthogonal click reactions (Figure 2). The phenotypic transformation of engineered non-polarized macrophages into the M1 subtype was activated by X-rays in vitro and confirmed in mice bearing 4T1 tumor xenografts, causing potent tumor-specific killing without signs of systemic toxicity.
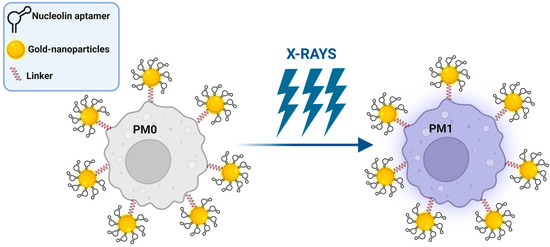
Figure 2. Schematic representation of PSA macrophage engineering strategy (see text for details). Created with BioRender.com (accessed on 2 March 2023).
These studies clearly show how the integration of aptamers in macrophage-guided immunotherapy is an effective strategy to enhance the antitumor effect of the IS.
4. Natural Killer Cells
NK cells are cytotoxic lymphocytes belonging to the innate IS, able to produce inflammatory cytokines and chemokines. They are called the “first line of defense” because, different from T lymphocytes, they do not express antigen-specific T cell receptors but act against mutated cells without prior sensitization or clonal expansion [42]. NK cell adoptive immunotherapy failed to show efficacy in the treatment of solid tumors, partly due to the immunosuppressive TME and lack of NK cell specificity to the tumor [[43].
Therefore, among the approaches for improving NK cell anticancer therapeutic efficacy, a great effort is focused on conferring cancer specificity through the expression of CARs or the conjugation of tumor-targeting ligands [44]. Zu and colleagues explored aptamers as active cancer-targeting agents by linking an aptamer, capable of specifically recognizing the CD30 receptor, on lymphoma cells to the surface of either an NK commercial cell line or NK cells obtained from three healthy donors [45]. This DNA-type aptamer was previously selected by the same group through a hybrid SELEX approach, in which steps of selection on CD30+ lymphoma cells were followed by selection steps on the CD30 recombinant protein [46]. The aptamer was modified at the 3′ end with lipophilic double C18 hydrocarbon chains for anchoring into the membrane of NK cells, which are so guided specifically to lymphoma cells to kill them [45]. More recently, the same authors applied the same approach in TNBC by attaching a DNA aptamer capable of binding a not-yet-known protein expressed on TNBC cells to the surface of NK cells. Aptamer-engineered NK cells inhibited lung metastasis from MDA-MB-231 cells intravenously injected in mice without side toxicity to normal tissues [47].
In order to further enhance the tumor-specificity of NK cells in solid tumors, dual aptamer-equipped NK cells were generated by using both an aptamer targeting hepatocellular carcinoma cells and the AptPD-L1 aptamer [28]. The resulting engineered NK cells were more effective than cells unconjugated or conjugated with only one of the two aptamers in inhibiting the growth of hepatocellular carcinoma in adoptively transferred mice. Another limit to the efficiency of immunotherapy with NK cells is their insufficient infiltration in solid tumors. Once again, aptamers have proved to be excellent tools for overcoming this problem. Hock’s group generated a bispecific aptamer-based conjugate capable of simultaneously binding to c-Met, a receptor highly expressed on several tumor cells, and to the Fcg receptor III (CD16a), a protein expressed on NK cells [48]. The conjugate is made up of the two highly specific c-Met and CD16a DNA aptamers that were fused by different linkers, preserving the ∼65 Å-ideal distance for simultaneously binding to the two receptors. The conjugate was able to efficiently mimic antibody-dependent cellular cytotoxicity by recruiting NK cells to cancer cells. Later, the same CD16 aptamer was fused to a PD-L1 DNA aptamer to generate a construct able to both recruit NK cells to PD-L1+ tumor cells and impair the PD-1/PD-L1 immunosuppressive axis by reactivating TILs against tumor cells in tumor-bearing mice [29]. This approach is particularly indicated for those solid tumors with high levels of PD-L1, such as TNBC.
References
- Park, J.H.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.B.; et al.et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. . Ann. Oncol. 2019, 30, 1941-1949.
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. . J. Immunother. Cancer 2016, 4, 59.
- Zheng, H.; Siddharth, S.; Parida, S.; Wu, X.; Sharma, D. Tumor Microenvironment: Key Players in Triple Negative Breast Cancer Immunomodulation.. Cancers 2021, 13, 3357.
- Zhao, Q.; Gong, Z.; Li, Z.; Wang, J.; Zhang, J.; Zhao, Z.; Zhang, P.; Zheng, S.; Miron, R.J.; Yuan, Q.; et al.et al. Target Reprogramming Lysosomes of CD8+ T Cells by a Mineralized Metal-Organic Framework for Cancer Immunotherapy. . Adv. Mater. 2021, 33, e2100616..
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications.. Front. Immunol. 2017, 8, 1850.
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al.et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370.
- Bertucci, F.; Finetti, P.; Birnbaum, D.; Mamessier, E. The PD1/PDL1 axis, a promising therapeutic target in aggressive breast cancers. Oncoimmunology 2015, 5, e1085148.
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742.
- Chen, C.; Li, S.; Xue, J.; Qi, M.; Liu, X.; Huang, Y.; Hu, J.; Dong, H.; Ling, K. PD-L1 tumor-intrinsic signaling and its therapeutic implication in triple-negative breast cancer. JCI Insight 2021, 6, e131458.
- Camorani, S.; Passariello, M.; Agnello, L.; Esposito, S.; Collina, F.; Cantile, M.; Di Bonito, M.; Ulasov, I.V.; Fedele, M.; Zannetti, A.; et al. Aptamer targeted therapy potentiates immune checkpoint blockade in triple-negative breast cancer. J. Exp. Clin. Cancer Res. 2020, 39, 180.
- Camorani, S.; Hill, B.S.; Collina, F.; Gargiulo, S.; Napolitano, M.; Cantile, M.; Di Bonito, M.; Botti, G.; Fedele, M.; Zannetti, A.; et al. Targeted imaging and inhibition of triple-negative breast cancer metastases by a PDGFRβ aptamer. Theranostics 2018, 8, 5178–5199.
- Camorani, S.; Hill, B.S.; Fontanella, R.; Greco, A.; Gramanzini, M.; Auletta, L.; Gargiulo, S.; Albanese, S.; Lucarelli, E.; Cerchia, L.; et al. Inhibition of Bone Marrow-Derived Mesenchymal Stem Cells Homing Towards Triple-Negative Breast Cancer Microenvironment Using an Anti-PDGFRβ Aptamer. Theranostics 2017, 7, 3595–3607.
- Passariello, M.; Camorani, S.; Vetrei, C.; Cerchia, L.; De Lorenzo, C. Novel Human Bispecific Aptamer-Antibody Conjugates for Efficient Cancer Cell Killing. Cancers 2019, 11, 1268.
- Passariello, M.; Camorani, S.; Vetrei, C.; Ricci, S.; Cerchia, L.; De Lorenzo, C. Ipilimumab and Its Derived EGFR Aptamer-Based Conjugate Induce Efficient NK Cell Activation against Cancer Cells. Cancers 2020, 12, 331.
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022.
- Camorani, S.; Granata, I.; Collina, F.; Leonetti, F.; Cantile, M.; Botti, G.; Fedele, M.; Guarracino, M.R.; Cerchia, L. Novel Aptamers Selected on Living Cells for Specific Recognition of Triple-Negative Breast Cancer. iScience 2020, 23, 100979.
- Camorani, S.; d’Argenio, A.; Agnello, L.; Nilo, R.; Zannetti, A.; Ibarra, L.E.; Fedele, M.; Cerchia, L. Optimization of Short RNA Aptamers for TNBC Cell Targeting. Int. J. Mol. Sci. 2022, 23, 3511.
- Camorani, S.; Tortorella, S.; Agnello, L.; Spanu, C.; d’Argenio, A.; Nilo, R.; Zannetti, A.; Locatelli, E.; Fedele, M.; Comes Franchini, M.; et al. Aptamer-Functionalized Nanoparticles Mediate PD-L1 siRNA Delivery for Effective Gene Silencing in Triple-Negative Breast Cancer Cells. Pharmaceutics 2022, 14, 2225.
- Agnello, L.; Tortorella, S.; d’Argenio, A.; Carbone, C.; Camorani, S.; Locatelli, E.; Auletta, L.; Sorrentino, D.; Fedele, M.; Zannetti, A.; et al. Optimizing cisplatin delivery to triple-negative breast cancer through novel EGFR aptamer-conjugated polymeric nanovectors. J. Exp. Clin. Cancer Res. 2021, 40, 239.
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248.
- Kim, M.; Lee, J.S.; Kim, W.; Lee, J.H.; Jun, B.H.; Kim, K.S.; Kim, D.E. Aptamer-conjugated nano-liposome for immunogenic chemotherapy with reversal of immunosuppression. J. Control. Release 2022, 348, 893–910.
- Somasunderam, A.; Thiviyanathan, V.; Tanaka, T.; Li, X.; Neerathilingam, M.; Lokesh, G.L.; Mann, A.; Peng, Y.; Ferrari, M.; Klostergaard, J.; et al. Combinatorial selection of DNA thioaptamers targeted to the HA binding domain of human CD44. Biochemistry 2010, 49, 9106–9112.
- Lai, W.Y.; Huang, B.T.; Wang, J.W.; Lin, P.Y.; Yang, P.C. A Novel PD-L1-targeting Antagonistic DNA Aptamer With Antitumor Effects. Mol. Ther. Nucleic Acids 2016, 5, e397.
- Wu, X.; Li, F.; Li, Y.; Yu, Y.; Liang, C.; Zhang, B.; Zhao, C.; Lu, A.; Zhang, G. A PD-L1 Aptamer Selected by Loss-Gain Cell-SELEX Conjugated with Paclitaxel for Treating Triple-Negative Breast Cancer. Med. Sci. Monit. 2020, 26, e925583.
- Li, T.; Yao, F.; An, Y.; Li, X.; Duan, J.; Yang, X.D. Novel Complex of PD-L1 Aptamer and Holliday Junction Enhances Antitumor Efficacy in Vivo. Molecules 2021, 26, 1067.
- Du, Y.; Zhang, D.; Wang, Y.; Wu, M.; Zhang, C.; Zheng, Y.; Zheng, A.; Liu, X. A highly stable multifunctional aptamer for enhancing antitumor immunity against hepatocellular carcinoma by blocking dual immune checkpoints. Biomater. Sci. 2021, 9, 4159–4168.
- Zhang, J.; Li, W.; Qi, Y.; Wang, G.; Li, L.; Jin, Z.; Tian, J.; Du, Y. PD-L1 Aptamer-Functionalized Metal-Organic Framework Nanoparticles for Robust Photo-Immunotherapy against Cancer with Enhanced Safety. Angew. Chem. Int. Ed. Engl. 2023, 62, e202214750.
- Zhang, D.; Zheng, Y.; Lin, Z.; Liu, X.; Li, J.; Yang, H.; Tan, W. Equipping Natural Killer Cells with Specific Targeting and Checkpoint Blocking Aptamers for Enhanced Adoptive Immunotherapy in Solid Tumors. Angew. Chem. Int. Ed. Engl. 2020, 59, 12022–12028.
- Zheng, A.; Du, Y.; Wang, Y.; Zheng, Y.; Ning, Z.; Wu, M.; Zhang, C.; Zhang, D.; Liu, J.; Liu, X.; et al. CD16/PD-L1 bi-specific aptamer for cancer immunotherapy through recruiting NK cells and acting as immunocheckpoint blockade. Mol. Ther. Nucleic Acids 2022, 27, 998–1009.
- Li, X.; Li, Z.; Yu, H. Selection of threose nucleic acid aptamers to block PD-1/PD-L1 interaction for cancer immunotherapy. Chem. Commun. (Camb.) 2020, 56, 14653–14656.
- Gao, T.; Mao, Z.; Li, W.; Pei, R. Anti-PD-L1 DNA aptamer antagonizes the interaction of PD-1/PD-L1 with antitumor effect. . Mater. Chem. 2021, 9, 746–756.
- Nakhjavani, M.; Shigdar, S. Future of PD-1/PD-L1 axis modulation for the treatment of triple-negative breast cancer. Pharmacol. Res. 2022, 175, 106019.
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61.
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237.
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 127.
- Ramesh, A.; Brouillard, A.; Kumar, S.; Nandi, D.; Kulkarni, A. Dual inhibition of CSF1R and MAPK pathways using supramolecular nanoparticles enhances macrophage immunotherapy. Biomaterials 2020, 227, 119559.
- Sylvestre, M.; Saxby, C.P.; Kacherovsky, N.; Gustafson, H.; Salipante, S.J.; Pun, S.H. Identification of a DNA Aptamer That Binds to Human Monocytes and Macrophages. Bioconjug. Chem. 2020, 31, 1899–1907.
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223.
- Sloas, C.; Gill, S.; Klichinsky, M. Engineered CAR-Macrophages as Adoptive Immunotherapies for Solid Tumors. Front. Immunol. 2021, 12, 783305.
- Qian, H.; Fu, Y.; Guo, M.; Chen, Y.; Zhang, D.; Wei, Y.; Jin, F.; Zeng, Q.; Wang, Y.; Chai, C.; et al. Dual-aptamer-engineered M1 macrophage with enhanced specific targeting and checkpoint blocking for solid-tumor immunotherapy. Mol. Ther. 2022, 30, 2817–2827.
- Chen, Y.; Gao, P.; Pan, W.; Shi, M.; Liu, S.; Li, N.; Tang, B. Polyvalent spherical aptamer engineered macrophages: X-ray-actuated phenotypic transformation for tumor immunotherapy. Chem. Sci. 2021, 12, 13817–13824.
- Wolf, N.K.; Kissiov, D.U.; Raulet, D.H. Roles of natural killer cells in immunity to cancer, and applications to immunotherapy. Nat. Rev. Immunol. 2023, 23, 90–105.
- Ghaedrahmati, F.; Esmaeil, N.; Abbaspour, M. Targeting immune checkpoints: How to use natural killer cells for fighting against solid tumors. Cancer Commun. (Lond.) 2023, 43, 177–213.
- Laskowski, T.J.; Biederstädt, A.; Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 2022, 22, 557–575.
- Yang, S.; Wen, J.; Li, H.; Xu, L.; Liu, Y.; Zhao, N.; Zeng, Z.; Qi, J.; Jiang, W.; Han, W.; et al. Aptamer-Engineered Natural Killer Cells for Cell-Specific Adaptive Immunotherapy. Small 2019, 15, e1900903.
- Parekh, P.; Kamble, S.; Zhao, N.; Zeng, Z.; Portier, B.P.; Zu, Y. Immunotherapy of CD30-expressing lymphoma using a highly stable ssDNA aptamer. Biomaterials 2013, 34, 8909–8917.
- Chen, Z.; Zeng, Z.; Wan, Q.; Liu, X.; Qi, J.; Zu, Y. Targeted immunotherapy of triple-negative breast cancer by aptamer-engineered NK cells. Biomaterials 2022, 280, 121259.
- Boltz, A.; Piater, B.; Toleikis, L.; Guenther, R.; Kolmar, H.; Hock, B. Bi-specific aptamers mediating tumor cell lysis. J. Biol. Chem. 2011, 286, 21896–21905.
More