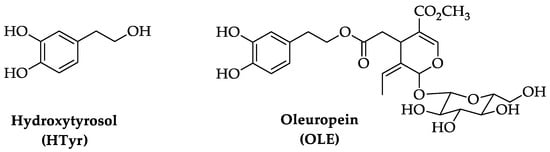
Aging is a multi-faceted process caused by the accumulation of cellular damage over time, associated with a gradual reduction of physiological activities in cells and organs. This degeneration results in a reduced ability to adapt to homeostasis perturbations and an increased incidence of illnesses such as cognitive decline, neurodegenerative and cardiovascular diseases, cancer, diabetes, and skeletal muscle pathologies. Key features of aging include a chronic low-grade inflammation state and a decrease of the autophagic process. The Mediterranean diet has been associated with longevity and ability to counteract the onset of age-related disorders. Extra virgin olive oil, a fundamental component of this diet, contains bioactive polyphenolic compounds as hydroxytyrosol (HTyr) and oleuropein (OLE), known for their antioxidant, anti-inflammatory, and neuroprotective properties.
- hydroxytyrosol and oleuropein chemistry
- pharmacokinetics
- brain neurodegeneration
1. Chemistry of Hydroxytyrosol (HTyr) and Oleuropein (OLE)
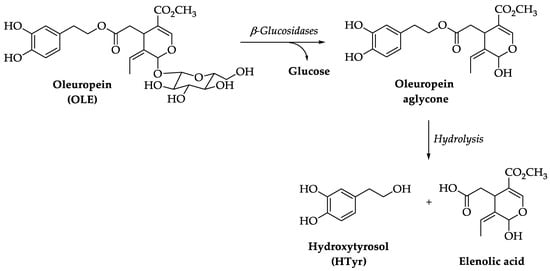
2. Pharmacokinetics of HTyr and OLE
2.1. Absorption
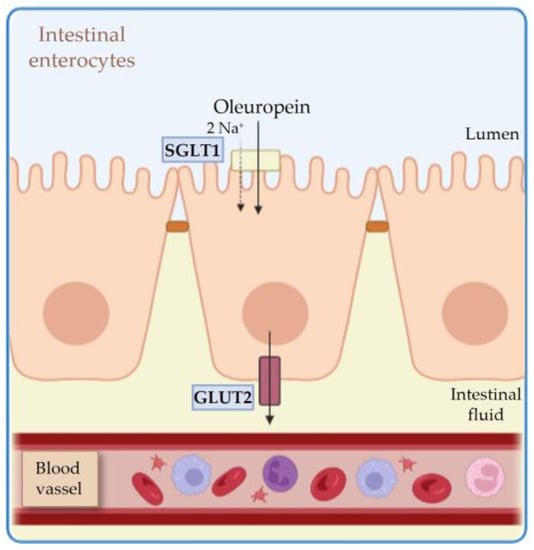
2.2. Metabolism and Distribution
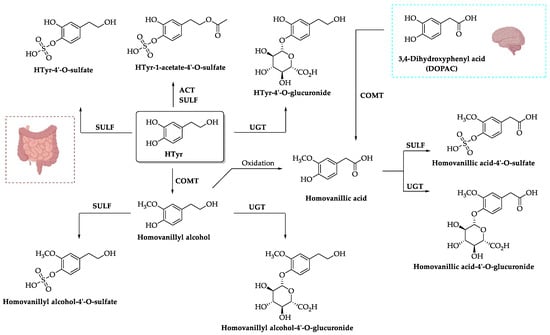
2.3. Excretion
2.4. Toxicity
3. Neuroprotective Function of HTyr and OLE in Neurodegenerative Diseases
3.1. Neuroprotective Function of HTyr and OLE in Alzheimer’s Disease
AD is a genetic and sporadic neurodegenerative disease that is a common cause of cognitive impairment acquired in mid- and late-life. AD is characterized by the formation of senile plaques and neurofibrillary tangles (NFT), constituted by extracellular deposit of β-amyloid (Aβ peptide) and by Tau protein accumulation, respectively [163][87]. In fact, the primary component of NFT are hyperphosphorylated Tau protein isoforms. The physiological function of Tau is to bind to microtubules, thus promoting their formation and stabilization. Microtubules are the main structural component of neurons and are responsible for axonal transport and axon growth. However, hyperphosphorylated Tau is unable to bind microtubules, whose stability is thus reduced, with consequent disruption of cellular traffic and synapse loss of function, cognitive decline, and dementia [163][87]. Several processes concur to the hyperphosphorylation of Tau, including Aβ peptides, impaired glucose metabolism, and inflammation [164][88]. Additionally, Aβ peptides are one of the main components of senile plaques and key responsible for AD pathogenesis; Aβ peptides derive from the amyloidogenic metabolism of amyloid precursor protein (APP), which is present in neurons and glia as well as in other tissues. The cleavage of APP by γ-secretase generates different peptides, of which Aβ (1–42) being hydrophobic tends to aggregate in plaques [165][89]. A remarkable effect of HTyr and OLE (of which HTyr is the main metabolite) is that they inhibit the fibrillization of Tau protein, thus decreasing the intraneuronal and glial lesions [127][90] (Daccache et al., 2011). Moreover, also β-amyloid aggregation is prevented by HTyr and OLE, since Aβ1–42 oligomer formation was shown to be inhibited in SH-SY5Y neuroblastoma cells [128,166][91][92] as well as in a mice model of Aβ deposition (adult TgCRND8 mice), where HTyr restored spatial and associative memory deficit [130][93]. Equivalent results were obtained after OLE administration in middle-aged TgCRND8 mice [131,132][94][95]. There is also evidence that treatment with HTyr reverted the deficit of spatial and working memory induced by intracerebroventricular injection in mice of Aβ1–42 oligomer and also prevented the activation of apoptotic pathways [129][96]. Reduced β-amyloid aggregation and increased lifespan by OLE was also observed in a transgenic C. elegans model expressing Aβ42 [133][97]. It is worth noting that OLE promotes insulin secretion in pancreatic β-cells and also prevents deposition of amylin in these cells through its HTyr metabolite [134][98]; similarly, in SH-SY5Y neuroblastoma cells OLE prevents the deposition of α-synuclein, another molecule in the path of β-amyloid aggregation [135][99]. Moreover, in the neural PC12 cell line, HTyr has been found to prevent the abnormal assembly of α-synuclein and to increase the expression of SIRT-2 deacetylase [136][100]. However, there is also evidence that the treatment of APP/Ps1 mice (a mouse model of AD) with a low dose of HTyr (5 mg/kg/day) for 6 months does not reduce Aβ deposition, although it is able to reduce mitochondrial protein oxidation and brain inflammation [137][101]. In comparison, the strong effects on Aβ deposition observed by Nardiello et al. (2018) were obtained with a shorter treatment at much higher dosage of HTyr, which suggests that the dosage is a key variable for effect, more than time [130][93]. Qin et al. (2021) showed also that HTyr treatment in the APP/PS1 mouse model significantly improved spatial memory in the Morris water maze test, reducing apoptosis in cortex and hippocampus [138][102]. Interestingly, this neuroprotective effect was dependent on the presence of estrogen receptor β (Erβ), which plays an important role as a neuroprotective agent [138][102]; in this regard, about two-thirds of AD patients are postmenopausal women. As mentioned in the Section 5, mitochondrial energetic deficit and altered energy metabolism are features of AD, and it turns out that HTyr is able to acutely induce mitochondria generation and fusion, in a model of Aβ toxicity and mitochondrial dysfunction (cell line 7PA2) [139][103]. Moreover, it has been shown that OLE and HTyr activate autophagy in SH-SY5Y cells exposed to toxic Aβ1–42 oligomers, thus preventing the accumulation of ROS and the impairment of mitochondrial function [140][104]. This is relevant considering that autophagy is the main cytoplasmic mechanism of removal and recycling of dysfunctional cellular components, and that the autophagic process decreases during aging, leading to the accumulation of toxic protein aggregates that can trigger neurodegeneration [167][105]. Another piece of evidence is a report showing that in N2a neuroblastoma cells HTyr or tyrosol counteracted cell death and the activation of NF-kB by nuclear translocation induced by treatment with Aβ25–35 oligomers. However, HTyr did not revert the decrease of glutathione (GSH) triggered by H2O2 or by Aβ [141][106]. Furthermore, HTyr neuroprotects astrocytes (C6 cell line) treated with Aβ25–35 oligomers by restoring insulin-signaling; this finding has neuropathological relevance, since astrocytes convert glucose into lactate, which is released into neurons for their functioning, and in fact insulin resistance occurring in diabetes doubles the risk of AD [142][107].3.2. Neuroprotective Function of HTyr and OLE in Parkinson’s Disease
PD is a 90% sporadic neurodegenerative pathology, affecting 1–2% of the population over the age of 65, characterized by a specific loss of dopaminergic neurons in the substantia nigra pars compacta [168][108]. PD is characterized by neuropsychiatric symptoms such as depression and anxiety that precede the onset of motor symptoms, consisting of resting tremors, muscular rigidity, and postural instability. Neuropathological hallmark is the presence of an intracytoplasmic inclusion body, known as a Lewy body, generated chiefly by abnormal aggregation of α-synuclein in substantia nigra, locus ceruleus, nucleus basalis, and hypothalamus [169][109]. α-synuclein, which is also a component of AD amyloid plaques (see above), is a presynaptic nerve terminal protein whose physiological function is neuroprotective and pro-neurogenic [170][110]. There is evidence that PD pathogenesis is associated to oxidative stress caused by ROS generated by a mitochondrial dysfunction in the respiratory chain [171][111]. It appears that in PD, etiology plays a role in the oxidation of dopamine by monoamine oxidase (MAO) in substantia nigra neurons, where unstable oxidized dopamine molecules (quinones) cause mitochondria dysfunction, formation of neurotoxic a-synuclein fibrils, dysfunction of the lysosomal system, and oxidative stress. ROS, in turn, activate microglial cells that promote neuroinflammation, causing death of dopaminergic neurons [172,173][112][113]. There are therefore several inter-related factors causing PD, while no specific therapy halting the neurodegenerative progression has been identified. In this context, HTyr butyrate has been shown to inhibit the apoptosis induced in SH-SY5Y cells by the PD-related neurotoxin 6-hydroxydopamine (6-OHDA) through activation of the Nrf2/heme oxygenase-1 axis (Nrf2/HO-1) [143][114]. Nrf2 is a transcription factor that plays a key role in the protection against oxidative stress, as it regulates a large number of antioxidant and anti-inflammatory genes and also modulates species longevity [174][115]. HTyr butyrate acts by inhibiting the ubiquitin E3 ligase that targets Nrf2 for proteasomal degradation, after reacting with the cysteine thiols of Kelch-like ECH-associated protein 1 (Keap1) that is part of the ubiquitin complex [143,175][114][116]. Consequently, Nrf2 is stabilized and accumulates in the nucleus where it induces the antioxidant enzyme HO-1 that, in turn, prevents cell damage by various types of ROS-dependent oxidative stresses [174][115]. Notably, Peng et al. (2015) [144][117] demonstrated that in PC12 cells the HTyr treatment causes a rapid increase of nuclear Nrf2 protein levels, which protects from death caused by H2O2 or 6-OHDA toxicity (see also below). It is worth noting that also in non-neural systems, HTyr has been found to activate HO-1 by stabilizing Nrf2, for example as part of a wound healing process (tested in vascular endothelial cell culture) or of a protective action against LPS-induced inflammation [176,177][118][119]. Moreover, in the Nrf2 and HO-1 activation by HTyr, the phosphatidylinositol 3-Kinase (PI3K)/Akt and extracellular signal-regulated kinase (ERK1/2) signaling pathways are also implicated, as assessed using specific inhibitors [176][118]; while the Janus kinase/Signal transducer and activator of transcription (Jak/Stat) pathway is inhibited by paracetylated HTyr [177][119]. As further example of Nrf2 induction by HTyr outside the nervous system, HTyr increased mRNA expression of Nrf2 and its downstream genes in the small intestine of diquat-challenged mice [178][120]. Additionally, OLE is able to neuroprotect from death PC12 cells exposed to 6-OHDA by reducing mitochondrial production of ROS and favoring autophagy [145][121]. In another work, Yu et al. (2016a) showed that treatment with HTyr protected from death SH-SY5Y cells exposed to dopamine or to 6-OHDA [146][122]. The authors proposed that this effect depends not only on HTyr antioxidant activity but also on its ability to induce phase II enzymes, including glutathione S-transferase (GST), HO-1, and NADPH quinone oxidoreductase 1 (NQO1), which lessens quinones’ toxicity by reducing them. Another interesting work shows that in PC12 cells, HTyr is able to inhibit the production of the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (DOPAL), generated in great amount from endogenous dopamine after inhibition of monoamine oxidase (MAO) through specific MAO inhibitors [147][123]. Therefore, HTyr was able to inhibit the oxidation of dopamine, either enzymatic or spontaneous, and this is relevant if researchers consider that MAO inhibitors are used for PD therapy. Another report shows that HTyr is able to acutely exert an antioxidant effect by inhibiting MAO enzymes after injection in mice striatum of 1-methyl-4-phenylpyridinium (MPP+), a potent oxidant [148][124]. As mentioned above, a theory for the etiology of PD is that the disease is caused by the increase of nigrostriatal dopamine catabolism by MAO enzymes, thus inducing a rise in ROS levels and death of dopaminergic neurons. A further report by the same authors [149][125] shows that pretreatment of rats with HTyr or with HTyr acetate or with nitro-HTyr 5 min before intrastriatal infusion of MPP+, protected from dopamine neuron degeneration, as ipsilateral turns were decreased and the GSH/GSSG ratio (glutathione/glutathione disulfide) was increased. Since GSSG is produced by antioxidant enzymes from GSH during the reduction of ROS, this indicates that the oxidative stress in the striatum was reduced. Additionally, OLE has been shown to be neuroprotective in a mouse model of PD induced by Rotenone, which inhibits the mitochondrial complex I causing overproduction of ROS and aggregation of α-synuclein. OLE (16 to 32 mg/kg) regulates the brain-derived neurotrophic factor (BDNF)/CREB/Akt signaling pathway, restoring BDNF levels and Akt phosphorylation and reducing aggregation of α-synuclein [150][126]. Remarkably, a biochemical study by Palazzi et al. (2020), conducted in the SH-SY5Y cell line, shows that HTyr did not change the naturally unfolded structure of α-synuclein but stabilizes certain areas of the molecule, thus preventing protein fibrillation [151][127]. Similarly, OLE was found to reduce the toxicity of α-synuclein aggregates favoring monomerization [152][128]. A further conformational study showed that OLE interacts with the N-terminal domain of α-synuclein, making it unable to react with cellular lipid membranes and thus preventing the generation of toxic aggregates [153][129]. Other studies in C. elegans showed how HTyr and OLE are able to increase lifespan in normal conditions or after heat stress and prevent α-synuclein aggregation, while in a PD model, HTyr increases locomotion and prevents α-synuclein accumulation in dopaminergic cells and in muscle, thus reducing neurodegeneration [154,155][130][131].3.3. Effects of HTyr and OLE in Diabetes-Induced Neurodegeneration
Type 1 and type 2 diabetes arise from a failure in insulin production or from resistance to insulin action, respectively. Insulin metabolism plays a major role in the onset of neurodegenerative diseases, in particular AD. Type 2 diabetes-induced hyperglycemia causes endothelial cells, pericytes, and astrocytes to enhance their mitochondrial respiration, which boosts ROS generation and oxidative stress. In fact, insulin receptors (Irs) are present in the brain, maximally in the cerebral cortex and hippocampus, and their ligands are insulin, IGF-1, and IGF-2 [179][132]. Through insulin and IGFs, Irs regulate the metabolism of glucose in the brain and exert inhibitory effects on apoptosis in brain neuronal cells [180][133]. The majority of cellular and molecular processes, including protein synthesis, sorting, transport, and degradation, as well as the maintenance of synaptic transmission, all require ATP, which can only be produced from glucose. Noradrenaline and/or cortisol, whose levels both increase with advancing age, can impede receptor activity at several locations, leading to desensitization of the neuronal insulin receptor in late-onset sporadic AD disease. Crucially, unregulated glucose metabolism causes AD symptoms [181][134]. In fact, insulin and amyloid-β compete for binding to IR. Insulin resistance is the outcome of this competition, which lowers the affinity of insulin to IR; thus, insulin resistance together with neuroinflammation and oxidative stress are causes of amyloid-β toxicity and accumulation of neurofibrillary tangles [182][135]. HTyr treatment of db/db mice, a model of type 2 diabetes, induces the Nrf2/HO-1 pathway in the brain and activates the PGC-1α transcriptional coactivator, AMPK kinase, and the deacetylase SIRT1 [156][136], which are part of an energy sensing network [183][137]. In fact, PGC1-α is, as mentioned in the Introduction, a master regulator of mitochondrial biogenesis and is directly regulated by the two metabolic sensors AMPK and SIRT1, through phosphorylation and deacetylation, respectively [183][137]. Thus, HTyr improves mitochondrial function and prevents oxidative stress in the db/db mice brain by activating the SIRT1/AMPK/PGC1α axis. Moreover, the amyloid protein (islet APP) generated in the diabetes mellitus pathology in β-cells of pancreas share a structure similarity with neuron APP and, consistently, islet APP fibrillation is prevented by HTyr [184][138]. It is worth noting that the activation of SIRT1 deacetylase by HTyr is functionally significant because SIRT1 exerts multiple actions that extend lifespan and have anti-aging effects [14][139]. These actions are indicated in the Introduction and include: (1) the inactivation of the pro-aging transcription factor NF-kB by deacetylating residue 310 of the p65/RelA subunit [185][140]; (2) blockage of the mTOR pathway, which results in rescue of the autophagy defect induced by oxidative stress [14][139]; notably, suppression of mTOR activity has been shown to increase lifespan in mice and other organisms [186][141]; (3) activation of PGC1α resulting in downregulation of ROS production mediated by NADPH-oxidase [187][142]; furthermore, PGC1α exerts an antisenescence action by promoting homeostasis of mitochondria, which, as mentioned above, is crucial to prevent oxidative stress, a condition that increases with aging. Another interesting study was conducted with OLE utilizing a streptozotocin (STZ)-based diabetic rat model. Authors observed that the outcome of diabetes consisted of reduced performance in spatial memory tests, neuroinflammation symptoms of oxidative stress such as decrease of superoxide dismutase (SOD), and of the pro-inflammatory cytokines IL-1β and TNF-α, together with a decrease of the phosphorylated forms of PI3K, AKT, and mTOR kinases in the hippocampus; all these changes were reverted by treatment with OLE, indicating that neuroinflammation and cognitive dysfunction can be attenuated by OLE [157][143]. More generally, there are several reports, reviewed by Zheng et al. (2021) [158][144], indicating that OLE displays multiple actions against diabetes, as it regulates insulin secretion, restores islet morphology, activates AMPK signaling, and ameliorates glucose tolerance and insulin resistance; consequently, OLE relieves diabetes-associated diseases including diabetic nephropathy, cardiovascular complications, and diabetic retinopathy [188][145].3.4. Effects of HTyr in Multiple Sclerosis
Multiple sclerosis (MS) is a chronic disease of the central nervous system characterized by loss of sensory and motor function, associated to immune-derived inflammation, demyelination, and consequent axonal damage. The clinical features indicate that MS patients frequently present recurrent episodes (relapses) of neurological disease, while for the majority (70%) the disease becomes chronic and progressive with age. Therefore, although MS is the most common cause of neurological disease in young adults (more than two million worldwide), the pathology aggravates during aging [189][146]. It is characterized by perivenular inflammatory lesions, caused by infiltrates of T-lymphocytes, leading to demyelinating plaques, which are the hallmark of MS. Inflammation results in oligodendrocyte damage and demyelination [190][147]. While axons remain almost intact during the early stages of the disease, the progressive evolution is an irreversible axonal damage [191][148]. The most common syndromes involve optic neuritis, brainstem, and spinal cord, and less frequently, parietal lobe syndromes. The inflammatory process in MS appears to be triggered by an autoimmune action implicating T-cells targeting myelin self-antigens, and to involve cross-reaction to antigens expressed by myelin elements or by viruses [192][149]. HTyr was shown to be able, in a rat experimental model of MS (experimental autoimmune encephalomyelitis, EAE), to reduce lipid and protein oxidation, and to increase GPX, thus reducing the oxidative stress produced by EAE [159][150]. Similarly, in primary rat astrocytes activated by LPS, HTyr, in a mix with tyrosol, inhibited matrix metalloproteinase-2 (MMP-2; also known as gelatinase A) and MMP-9 (gelatinase B); these are two proteolytic enzymes involved in inflammatory processes and in MS, since, for instance, MMP-9 increases blood–brain barrier permeabilization to leukocytes infiltration into the CNS [160][151]. This protective effect was not observed in treatment with the flavonoids quercetin and catechins. Additionally, OLE, administered by olive leaf extract (45 mg/kg) in an EAE rat model, up-regulated GPX1, SOD1 and SOD2 activity, SIRT1, and anti-inflammatory M2 microglia, whereas it downregulated proinflammatory M1, favoring myelin integrity. This indicates that OLE can be used to treat MS [161][152].3.5. Effects of HTyr in Huntington’s Disease
Huntington’s disease (HD) is a mostly hereditary neurodegenerative illness (autosomal dominant) caused by a mutation in the huntingtin protein, which results in an expanded number of the CAG trinucleotide repeat (i.e., over 28 repeats) within a polyglutamine tract in the molecule. Due to the multiple activities of huntingtin and to the cytotoxicity of large amounts of glutamine, this causes damage in basal ganglia (striatum) followed by degeneration in the cortex and hippocampus, accompanied by aberrant motor symptoms and depression [193][153]. HD onset depends on the extension of the mutation, but symptoms frequently appear during late age and worsen progressively [193][153].4. Effects of HTyr in Adult Neurogenesis and in Stroke
Adult neurogenesis is the process by which new neurons are generated throughout life in the brain from neural stem cells, in two neurogenic niches, the subgranular zone of the dentate gyrus of the hippocampus and the subventricular zone [194,195,196][154][155][156]. In the hippocampus, the new neurons are necessary for learning and memory, as they greatly enhance the built-in ability of the dentate gyrus to distinguish between similar memory patterns (pattern separation) [197,198][157][158]. The new neurons are continuously generated from stem cells in the subgranular zone of the dentate gyrus, which mature into proliferating progenitor cells and neuroblasts, before becoming post-mitotic neurons. There is debate about whether adult neurogenesis also occurs in humans, as there is evidence both in favor, even in old age [199][159], and against [200][160]. Moreover, the stem cell pool undergoes a slow process of self-renewal. Two models of self-renewal have been suggested, one proposing a recurrent self-renewal, where the same stem cell pool produces neurons through repeated rounds of division, remaining available for further activation [201[161][162][163][164],202,203,204], and another in which stem cells are rapidly depleted after a number of divisions [205][165]. In the first model, the pool is preserved also in old age. Notably, however, during aging there is a decrease in the generation of new neurons [206][166], which results in a reduced ability to perform hippocampus-dependent memory tasks [207][167]. Some neurogenic stimuli, such as physical exercise, can partially revert the decrease of neurogenesis occurring during aging [208[168][169][170],209,210], while other neurogenic stimuli, though effective in the adult, are unable to elicit neurogenesis in aged models. Between them are treatment with antidepressant molecules regulating 5-hydroxytryptamine (5-HT; selective 5-HT reuptake inhibitors, i.e., SSRI) or norepinephrine pathways [211[171][172][173],212,213], and learning [214,215][174][175]. Concerning nutrients, wresearchers have recently shown that HTyr activates hippocampal neurogenesis, including stem cells, in aged mice [47][24]. The majority of these neurogenic stimuli activates only progenitor cells and not stem cells, for example running [209][169] or the antidepressant fluoxetine [216,217,218][176][177][178]. However, some nutrients are also able to activate stem cells, though not in aged mice, but in models that are defective for neurogenesis, as for instance the natural flavonoid luteolin in a mouse model of Down syndrome [219,220][179][180]. Additionally, HTyr increases the survival of active (c-fos+) new neurons (see Section 10.3) and decreases the expression of the markers of aging and neuroinflammation lipofuscin and ionized calcium-binding adapter molecule 1 (Iba1) [47][24]. Overall, the finding that HTyr reactivates aging stem cells supports the idea of using dietary supplements to counteract cognitive deterioration during aging. Another report demonstrated that HTyr treatment of prenatally stressed rat mothers (by restraint stress on days 14–20 of pregnancy) restores in the offspring of treated mothers the mRNA levels of BDNF, GAP43, synaptophysin, and N-methyl-D-aspartate (NMDA) receptor subunits NR1, NR2A, and NR2B, all neural markers involved in synaptic plasticity and decreased by stress in the entire hippocampus [221][181]. However, no information was given regarding the generation of new neurons in the neurogenic niches [221][181]. More recently, a report about treatment with HTyr in vivo after stroke, obtained with transient occlusion of the middle cerebral artery, showed that HTyr-fed mice presented improved short-term recognition memory and an increase of BDNF and of cerebral blood flow in the hippocampus. Moreover, the study suggested a trend of increase of dentate gyrus total progenitor cells (DCX+ cells), without, however, showing a significant difference between treated and untreated conditions after stroke [222][182]. This may also depend on the dosage of HTyr treatment, lower than that used in the study by D’Andrea et al. (2020) [47][24]. This report, together with that by [221][181], brings to evidence the synaptogenic potential of HTyr, an important feature in the process of neural regeneration occurring after stroke. Additionally, OLE treatment, in a rat model of stroke by occlusion of the middle cerebral artery, induces a decrease of cerebral edema and plasma fibrinogen, inhibition of angiotensin converting enzyme (ACE) activity, and an increase of the antioxidant enzymes SOD, GPX, and catalase in brain tissue [223][183]. Another previous report in a mouse model of stroke by occlusion of the middle cerebral artery followed by reperfusion indicates that OLE (100 mg/kg i.p.) reduces the volume of cerebral infarction and apoptosis through reduction of Bax and increase of Bcl2 expression [224][184]. Likewise, OLE treatment after brain stroke caused in rats by intracerebral hemorrage following a collagenase injection in the brain improves brain edema and protects the integrity of the blood–brain barrier [225][185].5. Effects of HTyr and OLE in Stress, Anxiety and Depression-like Behavior
Depression is the most common psychiatric illness, affecting more than 350 million people worldwide. Depression is favored by stress, and its etiopathology has been related to defects in hippocampal neurogenesis, as the effect of antidepressants is conditional to their ability to induce neurogenesis in rodents as well as in non-human primates [211,217,218][171][177][178]. Stress and depression are associated with conditions that reduce the ability to generate new hippocampal neurons, such as high-plasma glucocorticoids, which inhibit neurogenesis; in fact, glucocorticoids and corticosterone are produced under stress in the adrenal gland, as part of the hypothalamic-pituitary-adrenal (HPA) axis, and glucocorticoid receptors are abundant in the hippocampus (see for review [235][186]. Conversely, inducers of hippocampal neurogenesis, such as antidepressants and voluntary exercise, reduce anxiety and depression-like behaviors induced by stress in experimental models, such as restraint, cold, or forced swimming. Indeed, in correlation to its pro-neurogenesis effects, running improves performance in the major tests assessing depression-like behavior (learned helplessness, forced-swim, and tail suspension) and anxiety (elevated plus-maze and open field) [236][187]. Remarkably, it has been shown that the optogenetic activation of the ventral dentate gyrus in the hippocampus, as detected by c-fos immunoreactivity, leads to a decrease of anxiety in mice, as measured by the plus maze test [237][188]. However, antidepressants and exercise exert antidepressant activity also when neurogenesis is prevented by chemical or genetic means; furthermore, the depletion of neurogenesis in itself is not sufficient to directly cause anxiety/depression [238][189], indicating that depression depends also on non-neurogenic factors, such as neural plasticity and blood vessel density. There is also a correlation between depression and aging-related processes, such as neuroinflammation, amyloid accumulation, changes in neuroplasticity, and synaptogenesis, that increase the risk of late-life-depression [239][190]. It is noteworthy that during aging, the production of new neurons decreases, and the antidepressant fluoxetine is unable to elicit neurogenesis in aged mice, but still has antidepressant activity through its modulation of neuronal plasticity, thus evidencing the importance of this component in depression [240][191]. In this context, it should be considered a potential antidepressant effect of HTyr, given its ability to stimulate hippocampal neurogenesis and neuron survival in young and aged mice [47][24]. In fact, a report showed that mice with depression-like behavior induced by chronic unpredictable mild stress (CUMS), after 7 weeks of treatment with HTyr, performed significantly better in forced swimming and tail suspension tests, in association with reduced oxidative stress—through enhanced SOD activity—and with increased number of glial fibrillary acidic protein (GFAP)-immunoreactive astrocytes, as well as increased activity of the BDNF/TrkB/CREB signaling pathway [226][192]. Considering that BDNF is important for the maturation of new neurons and is increased by neurogenic stimuli such as running and antidepressants [218[178][193],241], this report suggests that HTyr acts as antidepressant by stimulating neurogenesis. Another report using the CUMS model, explored the effect of HTyr on the hypothalamic–pituitary–adrenal (HPA) axis [227][194]. In fact, there is evidence that HPA axis is hyperactive in conditions of stress and in major depressive disorders [242][195] and contributes to the etiopathology. Fan et al. (2021) found that HTyr has a significant antidepressant effect associated to improvement of the HPA axis, as suggested by the decrease effected by HTyr after CUMS of serum corticosterone, adrenocorticotropic hormone (ACTH), and also TNF-α, IL1β, and IFN-γ [227][194]. However, they also observed in the hippocampus a strong HTyr-induced rescue of the decreased levels of 5-HT, which is a key player in the onset of depression. In fact, serotonin through hippocampal 5-HT1,3,4,6,7 receptors modulates proliferation [243][196] and the deletion of 5-HT1a receptor impairs the neurogenic effect of fluoxetine [211][171]. On the other hand, Fan et al. (2021) showed that HTyr was able to revert only partially the alteration in gut microbiota phyla [227][194]. Thus, HTyr displays a mixed antidepressant-like effect on the HPA axis and on the hippocampus. Similarly to HTyr, OLE treatment reduced in rats the anxiety elicited by a single prolonged stress (SPS), such as that induced by immobilization and forced swim tests, as assessed by the plus maze and open field tests that are able to detect the level of anxiety. The induction of a strong and prolonged stress, such as SPS, elicited symptoms similar to post-traumatic stress disorder (PTSD), a stress-related mental disorder caused by a traumatic experience [228][197]. A possible anti-PTSD mechanism of OLE is the ability to restore in the hippocampus the levels of Neuropeptide Y, which modulates the serotonergic pathways, as well as the levels of BDNF [228][197]. This is relevant considering the existence of stress-inhibitory pathways in the dentate gyrus activated by neurogenesis [237][188]. OLE (8 to 32 mg/kg i.p.) was also shown able to counteract depression-like symptoms elicited in mice by daily administration of corticosterone (40 mg/kg, i.p.) for 21 days. In fact, OLE treatment significantly improved the performance in the tail suspension test and in the forced swimming test, and restored brain serotonin and dopamine levels [229][198].6. Effects of HTyr and OLE in the Peripheral Nervous System: Nerve Damage, Neuropathies, and Regeneration
Older age increases the likelihood of hyperalgesia, as chronic pain incidence increases with age; in particular, peripheral neuropathic pain occurs in 35% of patients (typically due to diabetes or postherpetic neuralgia), and recovery from peripheral nerve damage takes longer [244][199]. In a report using a rat model of chronic compression of the dorsal root ganglion (CCD) to reproduce the neuropathic pain caused by intervertebral disc degeneration (IVDD), HTyr (acutely administered intrathecally in the spinal cord) reduced the levels of some inflammatory molecules activated by the NF-kB pathway, i.e., cyclooxygenase-2 (COX-2), NLRP3, nitric oxide synthase (iNOS), and metalloproteinase with thrombospondin motifs-4 (ADAMTS-4) [230][200]. Interestingly, another report demonstrated that HTyr (20 ng/mL) stimulates the proliferation of primary human Schwann cells as well as the protein expression of GFAP and p75 nerve growth factor receptor (p75 NGFR) [231][201]. Given that Schwann cells are implicated in peripheral nerve biology, including nerve formation and regeneration, transmission of nervous impulses along axons, and trophic support for neurons, this implies that the HTyr neuroprotective effects may facilitate regeneration and nerve trophism. Consistently, HTyr orally administered for 6 weeks (100 mg/Kg) was able to reduce peripheral neuropathy in STZ-treated diabetic rats [232][202]. In fact, HTyr counteracted the decrease of tail nerve conduction velocity and the increase of thermal nociceptive threshold occurring after STZ treatment. HTyr also abolished the decrease of Na+K+ ATPase activity in the sciatic nerve [232][202] indicating, as a whole, that HTyr may be a suitable therapy for the early-stage diabetic neuropathy. Additionally, OLE displays beneficial effects on inflammation ensuing spinal cord injury (SCI), since it was shown to reduce neutrophil infiltration, which plays a critical role in post-traumatic inflammation, as these cells cause large secondary tissue damage. Myeloperoxidase activity, a unique biomarker of the amount of neutrophil infiltration, was measured in order to determine if post-traumatic neutrophil infiltration had decreased [233][203]. HTyr treatment in vivo of rats (10 mg/Kg) also mitigated spinal cord injury after laminectomy, since the neural function was rescued and lipid peroxidation and myeloperoxidase activities were reduced, together with decrease of proinflammatory cytokines and apoptotic markers [234][204].7. HTyr and OLE in Senescence and Lifespan
7.1. Effects of HTyr and OLE on Senescence
Cellular senescence is characterized by a permanent arrest of cell proliferation due to stress conditions, including telomere shortening, oxidative stress, hypoxia, oncogenic activation, and DNA damage, and has been linked to processes such as tumor suppression, tissue repair, embryogenesis, and aging. The principal hallmarks of senescence are an increase in senescence-associated βgalactosidase activity (SA-β Gal), the presence of telomere dysfunction-induced foci (TIF), upregulation of specific cell cycle regulators (mainly the p53-p21 and p16-pRb axes), altered gene expression patterns, and the activation of a senescence-associated secretory phenotype (SASP), implying the synthesis and secretion of inflammatory mediators, growth factors, and extracellular matrix proteins [270,271][205][206]. The SASP is thought to play a major role in many age-related diseases, such as AD [272][207] and cancer [273][208], where it contributes to the maintenance of chronic inflammation, and in cardiovascular diseases and type 2 diabetes, also characterized by a low-grade chronic inflammation state. It is worth noting that the elimination of senescent cells in progeroid and aged mice, through senolytic drugs or genetic mutations, markedly improves health span after senescent cells are cleared [270,274][205][209]. Several papers have reported that HTyr and OLE are endowed with the ability to modulate senescence or related inflammation in primary mammalian cell cultures, which represent suitable cellular models to study in vitro the mechanisms of aging. In pre-senescent human lung cells (MRC5) and neonatal human dermal fibroblasts (NHDFs), chronic (4–6 weeks) treatment with 1 μM HTyr or 10 μM OLE aglycone reduced some known markers of senescence, such as p16 and SA-β Gal. Moreover, the treatment decreased the secretion of senescence/inflammation markers such as IL-6 and metalloprotease, and levels of COX-2 and α-smooth-actin. Furthermore, the induction of inflammation following exposure to TNFα was abolished in OLE- and HTyr-pre-treated NHDFs [275][210]. Thus, the modulation of the senescence and senescence-associated inflammatory phenotype might be an important mechanism underlying the beneficial effects of olive oil phenols. Moreover, OLE (5 μM) and HTyr (1 μM) exerted a protective effect on 8 Gy irradiation-induced senescence, in γ-irradiated NHDFs, mitigating DNA damage and reducing the SASP mediated by the cyclic GMP AMP synthase/stimulator of intereferone genes (cGAS/STING)/NFκB pathway [276][211]. The ability of OLE and HTyr to alleviate senescence status, and the related SASP in normal cells can be exploited to improve the efficacy and safety of cancer radiotherapy. Similar results were obtained in human dermal fibroblasts (HDFs) exposed to UVA. In this cell line, HTyr decreased SA-β-galactosidase activity and expression levels of MMP-1 and MMP-3 in a dose-dependent manner. These observations were accompanied by an anti-inflammatory effect, demonstrated by the reduced expression of IL-1β, IL-6, and IL-8 genes [277][212]. Interestingly, Varela-Eirín et al. (2020) demonstrated that OLE has senolytic activity in osteoarthritic chondrocytes (OACs) [278][213]. In fact, OLE reduced the inflammatory and catabolic factors mediated by NF-kB (IL-1ß, IL-6, COX-2, and MMP-3) and lowered cellular senescence in OACs, synovial and bone cells.7.2. Effects of HTyr and OLE on Lifespan
Proliferative cellular lifespan is a complex process that is governed by multiple pathways in part interdependent with cellular senescence. In fact, normal somatic cells in vitro, after a finite number of cell divisions, can enter in the non-proliferative state of senescence [279][214]. In in vitro models, it was observed that treatment with OLE extended the lifespan of normal human fibroblasts (NHFs) by enhancing the activity of proteasome [280][215], which is known to decline during aging in human tissues as well as in senescent primary culture cells. Moreover, HTyr treatment was able to increase chronological lifespan (CLS) in quiescent NHFs, which have lost their capacity to repopulate. CLS measures the length of time nondividing cells survive before reach senescence, without telomere attrition. This process could be related to the observation that quiescent human somatic cells exhibit age-dependent loss in their regenerative capacity. The observed HTyr-induced extension of CLS was associated with an increase in manganese superoxide dismutase (SOD2) activity and reduction of age-associated mitochondrial ROS generation [281][216]. It is noteworthy that, as mentioned in previous sections, HTyr and OLE have shown the ability to activate AMPK and SIRT1 pathways and inhibit the mTOR and insulin/IGF1 signaling, which are crucially involved in regulation of health span and, as a consequence, lifespan and senescence in animal models. Interestingly, numerous studies have been focused on the effects of HTyr and OLE on the lifespan of Caenorhabditis elegans. This free-living nematode is a powerful animal model mostly used in aging research due to its short life cycle. It was recently observed that OLE could significantly prolong the lifespan of C. elegans increasing the survival rates of worms against lethal heat shock and oxidative stress. OLE supplementation increased the expression and activity of antioxidant enzymes and suppressed the generation of malondialdehyde in nematodes. These positive effects on longevity and stress resistance were mediated by the factors DAF-16/FoxO, which plays a vital role in the insulin/IGF-1 signalling (IIS) pathway, and the SKN-1/Nrf2 pathway, another stress response and longevity pathway [282][217]. HTyr was also able to increase the survival of worms after heat stress and could further prolong the lifespan in unstressed conditions. In addition, in aged worms, exposure to HTyr and OLE led to the improvement of locomotive behavior and the attenuation of autofluorescence as a marker of ageing [154][130]. Moreover, numerous studies have been conducted on the beneficial effects of olive oil polyphenols in C. elegans models of PD and AD. Although C. elegans is not able to develop PD, exposure to the pesticide rotenone or the transgenic expression of human α-synuclein induces the Parkinsonian-like syndrome in C. elegans, which manifests as impaired movement. In models of PD, HTyr and OLE, as already mentioned in Section 6.2, have been shown to enhance swim performance of worms, to reduce α-synuclein accumulation in muscle cells, and to prevent neurodegeneration in α-synuclein-containing dopaminergic neurons [154][130]. Analogously, the treatment with a natural extract enriched in HTyr showed beneficial effects on the lifespan of wild-type nematodes and in a PD-model [155][131]. More recently, it was observed that in C. elegans, treatment with an olive fruit extract 20% rich in HTyr had beneficial effects on AD features such as Aβ- and tau-induced toxicity, as well as on oxidative stress. The treatment prevented oxidative stress and delayed Aβ-induced paralysis reducing Aβ aggregates. Indeed, the extract partially reduced the proteotoxicity associated with aggregation of the tau protein. The observed effects were correlated to the activity of the SKN-1/NRF2 transcription factor and the overexpression of HSP-16 [283][218].8. Protective Effects of HTyr and OLE against Skeletal Muscle Dysfunction
In addition to its fundamental function in locomotion and maintenance of posture, skeletal muscle plays crucial roles in energy and glucose metabolism. This tissue has also emerged as an endocrine organ producing and releasing growth factors and cytokines (commonly termed myokines) that modulate systemic physiology. Furthermore, skeletal muscle may cross-talk with the nervous system and other tissues via direct muscle–nerve interactions, release of metabolites, systemic adaptation deriving from the energy demand of contracting muscle (e.g., during exercise), and myokines [21,284][219][220]. The age-associated loss of muscle mass and strength (called sarcopenia) is an important medical condition that results in fragility and reduced mobility and negatively affects bone mass, which also declines during aging causing osteopenia and osteoporosis. The concomitance of sarcopenia and osteoporosis in elderly patients leads to severe physical disability, frailty, high risk of falls, and osteoporotic fractures [285][221]. Furthermore, sarcopenia also influences global metabolic homeostasis, lifespan, systemic aging, and progression of age-related degenerative diseases in non-muscle tissue, as well as the organism’s responses to oxidative stress and dietary restriction [286][222].8.1. Age-Related Sarcopenia and Osteoporosis
Multiple factors contribute to sarcopenia of aging, including chronic activation of inflammatory pathways, enhanced ROS generation and/or dysregulation of redox signaling, loss of muscle proteins resulting from an imbalance between protein synthesis and degradation, dysregulation of muscle autophagy, and impairment of neuromuscular transmission. These age-associated dysfunctions lead to myofiber death, muscle atrophy, and reduced regenerative potential [287,288][223][224]. A few studies have shown beneficial effects exerted on sarcopenia by olive mill wastewaters- and olive leaf-derived extracts rich in HTyr and/or OLE. Pierno and colleagues (2014) tested the therapeutic potential of a polyphenolic mixture (LACHI MIX HT), extracted from olive mill wastewaters and containing mainly HTyr and low amounts of other phenolic compounds (tyrosol, catechol, gallic acid, homovanillic acid, and caffeic acid) in the amelioration of skeletal muscle dysfunctions due to aging-associated oxidative stress [289][225]. The effects of treatment of old rats with LACHI MIX HT were compared with those of purified HTyr. LACHI MIX HT treatment was able to counteract some of the alterations in excitation/contraction function typically observed in aged skeletal muscle. In particular, the treatment was shown to increase muscle weight, to improve sarcolemma chloride channel conductance, contractile function, and ATP-dependent potassium channel activity and to decrease blood creatine kinase levels and the resting cytosolic calcium concentration. Furthermore, LACHI MIX was more effective than purified HTyr in ameliorating most of the examined parameters, possibly due to the synergic activity of its various components. Extracts from olive tree (Olea europaea) leaves are rich in OLE and HTyr, have a high antioxidant and scavenging power, and have been reported to exert beneficial effects on several health conditions, including hypertension, cardiovascular diseases, diabetes, and hyperlipidemia [290][226]. A recent study analyzed the effects of treatment with an olive leaf extract on aging-related sarcopenia and skeletal muscle insulin resistance [291][227]. Sarcopenia is known to be associated with: (i) increased expression of the E3 ubiquitin ligases Muscle Ring-finger protein-1 (MURF1) and Atrogin-1, which play a major role in the process of age-induced muscle loss [292][228]; (ii) increased expression of myostatin, a secreted myokine that inhibits skeletal muscle growth and negatively impacts muscle metabolism [293,294][229][230]; (iii) upregulation of histone deacetylase-4 (HDAC4), a deacetylase that promotes muscle atrophy in response to multiple stimuli by targeting several substrates, including myosin heavy chains (MyHC) and the transcriptional regulator PGC1α [295][231]; (iv) upregulation of the myogenic regulatory factors MyoD and myogenin, possibly as a compensatory mechanism [296][232]. A 21-day treatment of old rats with an olive leaf extract was shown to attenuate the age-induced atrophy of gastrocnemius muscle and the alterations in the mRNA expression of many of the above- mentioned markers of sarcopenia. Specifically, the authors observed a reduction of HDAC4, MyoD, myogenin, myostatin levels, and upregulation of PGC1α levels. The treatment also prevented the age-induced elevation of several pro-inflammatory markers and enhanced the expression of the anti-inflammatory cytokine IL-10 that was reduced in old untreated animals. Furthermore, the treatment improved muscle insulin sensitivity in gastrocnemius muscle by activating the insulin-dependent PI3K/Akt pathway [291][227]. In a follow-up study, the same authors evaluated whether the addition of the olive leaf extract to an oil mixture, composed of 25% algae oil and 75% EVOO (AO:EVOO), could synergistically attenuate sarcopenia in old rats [297][233]. In previous studies, the AO:EVOO mixture per se was reported to exert protective effects against cardiometabolic and skeletal muscle alterations associated with aging [298,299][234][235]. Combined treatment of 24-month-old rats with the olive leaf extract and AO:EVOO mixture provided further benefit to sarcopenia preventing not only the decrease in the weight of gastrocnemius muscle but also in the weight of the soleus muscle. Besides preventing the induction of the sarcopenia-related markers HDAC4, MyoD, myogenin, and rescuing the downregulation of PGC1α, the co-administration of both ingredients attenuated the reduction of MyHC isoforms, downregulated the expression of Atrogin-1 and MuRF1, prevented age-induced macrophage infiltration, and activated the insulin-dependent PI3K/Akt pathway in gastrocnemius muscle and adipose tissue [297][233]. There is ample evidence for a constant cross-talking between skeletal muscle and bone via paracrine and endocrine signals and for the existence of common pathogenic pathways shared by sarcopenia and osteoporosis including oxidative stress, increased inflammatory cytokine production, reduced anabolic hormone secretion, and reduced physical activity [300,301][236][237]. Consumption of olives, olive oil and olive polyphenols has been shown to improve bone health [302,303][238][239]. In particular, a number of studies reported physiological effects exerted by HTyr and/or OLE against osteoporosis. Hagiwara et al. (2001) evaluated the effects of HTyr and OLE on bone formation using cultured osteoblasts and osteoclasts, and on bone loss in ovariectomized mice, a widely used experimental model of post-menopausal osteoporosis [304][240]. They showed that HTyr and OLE inhibited the formation of multinucleated osteoclasts in culture, and that OLE enhanced the deposition of calcium by osteoblasts. Furthermore, OLE and HTyr were able to decrease bone loss in trabecular bone of femurs in ovariectomized mice. Other studies reported that OLE, HTyr, and olive mill wastewater extracts elicited beneficial effects on femur bone mineral density and improved inflammatory and oxidative stress markers in an experimental model of senile osteoporosis induced by ovariectomy associated with an acute inflammation in rats [305,306][241][242]. More recently, a beneficial effect of OLE on bone mineral density of lumbar vertebra and left femur of ovariectomized rats was also reported by Liu et al. (2022) [307][243]. Moreover, OLE could reduce the serum levels of IL-6 and malondialdeyde (a marker of lipid oxidation). In cellular experiments, it could also promote the proliferation of primary osteoblasts while inhibiting osteoclast differentiation by upregulating expression of osteoclastogenesis inhibitory factor (OPG) and downregulating expression of receptor activator for nuclear factor-κB ligand (RANKL). Finally, a study conducted in the murine osteoblast MC3T3-E1 cell line revealed a cytoprotective effect of HTyr against oxidative stress-induced osteoblast apoptosis and shed light on the underlying mechanism [308][244]. In fact, HTyr was shown to prevent oxidative stress-induced mitochondrial dysfunction and osteoblast apoptosis by decreasing the cleavage of optic atrophy 1 (OPA1), one of the most important mitochondrial dynamics proteins, and by activating the Akt/glycogen synthase kinase 3 β (GSK3β) signaling pathway.8.2. Skeletal Muscle Atrophy and Oxidative Stress
In addition to aging, skeletal muscle atrophy is the consequence of various conditions, such as muscle disuse, strenuous exercise, denervation, neurodegenerative disease, muscular dystrophies, obesity, and diabetes. Periods of immobilization are often associated with aging. Prolonged periods of muscle inactivity result in oxidative stress and chronic elevation of ROS production, primarily derived from mitochondria, within inactive muscle fibers. Such disturbed redox signaling critically contributes to disuse-induced muscle atrophy through several mechanisms, including (i) inhibition of muscle protein synthesis via repression of the anabolic Akt/mTOR signaling; (ii) activation of the Ca2+-activated proteases calpains; (iii) acceleration of protein degradation via the ubiquitin–proteasome system; (iv) activation of apoptosis; (v) stimulation of autophagy [309][245]. By using a rat model of unloading-induced muscle atrophy (3-week tail suspension), Liu et al. (2014) showed that 7 days’ reloading efficiently rescued skeletal muscle atrophy and mitochondrial dysfunctions [310][246]. Interestingly, administration of a mixture of mitochondrial nutrients including HTyr, α-lipoic acid, acetyl-L-carnitine, and coenzyme Q10 for 4 weeks to unloaded rats exerted a reloading-like effect and promoted the recovery initiated by reloading. Specifically, treated animals showed enhanced motor function, increased soleus muscle weight, decreased protein degradation, and apoptosis. Furthermore, the nutrient mixture rescued unloading-induced mitochondrial defects, by increasing total anti-oxidative capability, mitochondrial copy number and electron transport chain complex I and II activities and markers of mitochondrial biogenesis, such as PGC1α, nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (Tfam). Redox-sensitive signaling pathways also play a pivotal role in exercise-mediated remodeling of skeletal muscle. Skeletal muscle shows different responses to the type of exercise, as well as its frequency, intensity, and duration. Increased ROS production in contracting muscle fibers plays a required role in skeletal muscle adaptations induced by regular endurance or resistance exercise training, while excessive ROS produced by strenuous or acute exercise can cause muscle oxidative stress, fatigue, and damage [309,311,312][245][247][248]. As reported by Feng et al. (2011), an 8-week intensive exercise program in rats resulted in decreased endurance capacity and highly induced expression of muscle atrophy (MURF1 and Atrogin-1) and autophagy markers (Atg7, Beclin-1 and LC3) [313][249]. This was accompanied by increased mitochondrial fission induced by excess of ROS in skeletal muscle and a decrease in PGC1α and complex I subunit expression. All of these changes were eliminated by treatment of exercised rats with HTyr. In addition, HTyr enhanced mitochondrial fusion and mitochondrial complex I and II activities and inhibited the expression of the oxidative-stress-responsive proteins p53, p21, and SOD2 in muscles of exercised rats [313][249]. Therefore, HTyr appears to improve exercise capacity by protecting contracting muscles from excessive ROS production.8.3. Oxidative Damage and Skeletal Muscle Cell Degeneration
Protective effects of HTyr and HTyr derivatives on oxidative stress- and inflammation-induced mitochondrial dysfunction and muscle cell degeneration were also described in the C2C12 myogenic cell line. Wang et al. (2014) reported that induction of oxidative stress in C2C12-differentiated myotubes promoted significant mitochondrial dysfunction in a time-dependent manner, accompanied by decreased expression of MyHC and myogenic regulatory factors (MyoD, myogenin, MRF4) and induction of apoptosis [314][250]. Furthermore, oxidative stress was shown to promote rapid cleavage of OPA1, a protein that controls mitochondrial inner membrane fusion and remodeling. Pre-treatment with HTyr acetate, which is present in olive oil at a concentration similar to that of HTyr, significantly prevented oxidative stress-induced OPA1 cleavage and mitochondrial morphology changes, and this was accompanied by improvement of mitochondrial oxygen consumption capacity, ATP productive potential and activities of mitochondrial complexes I, II, and V, and inhibition of oxidative stress-induced MyHC decrease [314][250]. Another study showed that pre-treatment with HTyr or the corresponding ester HTyr laurate effectively protected C2C12 myoblasts from apoptotic cell death induced by H2O2 treatment [105][82]. Furthermore, pre-treatment with HTyr was shown to counteract muscle cell degeneration induced by the inflammatory cytokine TNFα in C2C12 differentiating myoblasts by increasing the expression of differentiation markers (MyHC and myogenin), PGC1α and mitochondrial complexes I and II, as well as the activity of muscle creatine kinase [315][251]. Finally, a couple of studies investigated the effects of OLE in primary avian skeletal muscle cells. Kikusato and colleagues (2016) reported that treatment of avian muscle cells with OLE induced the mRNA expression of avian-specific uncoupling protein (avUCP), PGC1α, and downstream mitochondrial biogenesis-related genes (NRF1, Tfam, ATP5a1), as well as the activity of mitochondrial cytochrome c oxidase (COX) [316][252]. Additionally, the expression of the SIRT1 deacetylase was found upregulated by OLE. Since it is well-known that deacetylation by SIRT1 induces the PGC1α co-transcriptional activity [181][134], these results suggested that OLE upregulates mitochondrial biogenesis and avUCP expression in muscle avian cells through the SIRT1/PGC1α regulatory axis. Furthermore, OLE was observed to suppress the levels of mitochondrial ROS generation, possibly via the up-regulation of SOD2 and avUCP gene expression. In a follow-up study, Muroi et al. (2022) used a specific inhibitor of SIRT1 activity to confirm that indeed it can mediate the enhancement of PGC1α, avUCP, ATP5a1, and SOD2 gene expression and the suppression of mitochondrial ROS generation induced by OLE in chicken muscle cells [317][253]. The study also showed that the action of OLE on the above markers of mitochondrial biogenesis and oxidative damage can be mediated by transient receptor potential cation channel subfamily V member 1 (TRPV1), as determined by using an antagonist of TRPV1. Furthermore, treatment of chicken muscle cells with OLE was shown to increase intracellular Ca2+ concentration and stimulate the phosphorylation, and hence activation, of the AMPK kinase in a TRPV1-dependent manner. Based on the knowledge that Ca2+/calmodulin-dependent protein kinase 2 induces phosphorylation of AMPK and that AMPK, SIRT1, and PGC1α are part of a highly coordinated regulatory network [181[134][254],318], the authors proposed that the effects of OLE in chicken muscle cells may be due to induction of Ca2+ influx, possibly through the activation of TRPV1 localized at ER, and increased activity of AMPK, followed by SIRT1 activation and then activation of PGC1α and induction of downstream mitochondrial targets [317][253].8.4. Skeletal Muscle Insulin Resistance and Metabolic Syndrome
As mentioned earlier, skeletal muscle plays a crucial role in whole-body energy homeostasis and is a primary tissue of insulin-induced glucose uptake and oxidative metabolism. Muscle atrophy and sarcopenia are associated with the development of skeletal muscle insulin resistance [319][255], which is considered to be a major driver of metabolic syndrome (MetS), a cluster of medical conditions (obesity, hypertension, dyslipidemia, hyperglycemia) that together increase the risk of developing type 2 diabetes, cardiovascular diseases, and stroke. Major factors in the development of MetS include sarcopenia, chronic inflammation, abdominal obesity, insulin resistance/hyperinsulinemia, physical inactivity, high fat intake, and genetic factors. MetS, obesity, and diabetes induce adverse effects on skeletal muscle function, including muscle fiber atrophy and contractile dysfunctions, altered metabolism, insulin resistance, oxidative stress, mitochondrial dysfunctions, and reduced regenerative potential [319,320,321,322,323][255][256][257][258][259]. Commonly used MetS murine models exhibit metabolic disorders induced by high-fat diet (HFD) feeding or a non-functional leptin pathway, such as the db/db mouse [328][260]. Cao and colleagues extensively examined the effects of HTyr in skeletal muscle and liver of C57BL/6 mice fed a HFD with or without supplementation of low-dose or high-dose HTyr (10 or 50 mg/Kg/day, respectively) for 17 weeks [329][261]. HTyr administration was shown to effectively inhibit body and organ weight increase (50 mg/Kg/day). Both low- and high-dose HTyr treatment enhanced glucose tolerance and lowered the serum levels of glucose, insulin, lipids, and inflammatory cytokines. In skeletal muscle and liver, HTyr reduced the accumulation of lipid deposits through inhibition of the sterol regulatory element-binding transcription factor (SREBP) pathway, attenuated oxidative stress by enhancing antioxidant enzyme activity, elevated the decreased expression of complex I and II subunits of the electron transport chain induced by HFD, and lowered the expression of markers of mitochondrial fission and apoptosis. Moreover, in muscle tissue, HTyr decreased the level of mitochondrial protein carbonylation and elevated the activities of complexes I, II, and IV. The effects of HTyr were also examined in diabetic db/db mice treated for 8 weeks with low-dose HTyr (10 mg/Kg/day) or with metformin, a commercial antidiabetic drug, as a positive control. Both HTyr and metformin reduced the fasting blood glucose levels in db/db mice, whereas the fasting serum levels of lipids (triglyceride, cholesterol) were lowered by HTyr but not by metformin. Furthermore, HTyr was shown to be more effective than metformin in reducing protein and lipid damage in skeletal muscle and liver and in increasing the activities of mitochondrial complexes I and IV [329][261]. Another study evaluated the effects of HTyr acetate (HTyr-Ac) on glucose consumption in C2C12 skeletal muscle cells and 3T3-L1 adipocytes. Treatment with increasing concentration of HTyr-Ac was reported to stimulate glucose uptake in a dose-dependent manner in both C2C12 myotubes and differentiated adipocytes. Furthermore, HTyr-Ac was shown to exert an anti-adipogenic effect by inhibiting adipocyte differentiation and by stimulating lipolysis of fully differentiated adipocytes [330][262]. With regard to OLE, it has been shown that C57BL/6 mice fed an HFD supplemented with 0.038% of OLE, for 12 weeks, exhibited reduced levels of fasting glucose and improved insulin resistance. Moreover, the gastrocnemius muscle of OLE-fed mice displayed increased expression and membrane localization of glucose transporter type 4 (GLUT4) protein [331][263]. In the same study, C2C12 myotube cells treated with OLE displayed enhanced glucose uptake and GLUT4 translocation to the plasma membrane. Furthermore, OLE improved insulin sensitivity in C2C12 cells treated with palmitic acid, a model of lipotoxicity and insulin resistance. OLE did not show a synergistic effect with insulin regarding glucose uptake, and stimulated phosphorylation, and hence activation, of AMPK but not phosphorylation of the Akt kinase, which is downstream of the insulin signal [331][263]. Therefore, the mechanism through which OLE can induce glucose uptake in muscle cells appears to be linked to the activation of AMPK but does not require activation of the PI3K/Akt pathway. Similar results were reported by Hadrich et al. (2016) who showed that treatment of C2C12 myotubes with OLE can stimulate glucose uptake and activate AMPK and MAPK signaling but not the PI3K/Akt insulin signaling pathway [332][264]. In addition, the treatment was shown to protect C2C12 myotubes against oxidative stress induced by H2O2 by decreasing ROS production and lipid peroxidation levels. In agreement with the previously discussed studies, Alkhateeb et al. (2022) have recently reported protective effects of OLE on muscular insulin resistance in isolated soleus muscle preparations treated for 12 h with a high concentration of palmitate [333][265]. In fact, OLE treatment enhanced insulin-stimulated glucose uptake, the translocation of GLUT4 at the plasma membrane, and the levels of Akt substrate of 160 kDa (AS160) phosphorylation. Furthermore, OLE promoted the activation of AMPK and an inhibitor of AMPK blocked OLE-stimulated glucose uptake, GLUT4 translocation, and AS160 phosphorylation. This indicated that OLE can ameliorate palmitate-induced insulin resistance via an AMPK-dependent mechanism. As mentioned earlier, OLE is the most abundant phenolic compound in extracts from olive leaves. By using a rat model of STZ-induced diabetes, Giacometti et al. (2020) investigated the effects of an OLE-rich olive leaf extract on GLUT4 expression and intracellular vesicular GLUT4 trafficking in soleus muscle [334][266]. Diabetic rats treated for 10 days with 512, 768 or 1024 mg/Kg of olive leaf extract (containing 20.3, 33 or 44.5 mg/kg of OLE, respectively) displayed significantly reduced blood levels of glucose and triglycerides. Histopathological examination of the diabetic soleus muscle revealed a decrease in fiber size and increased fibrosis, and such pathological muscle alterations were improved after treatment with the olive leaf extract in a dose-dependent manner. Furthermore, treatment with olive leaf extract was shown to promote GLUT4 translocation to the myofiber membrane in soleus muscle and to enhance the expression levels, and the colocalization with GLUT4, of the Rab GPTases Rab8A, Rab13, and Rab14, which are central regulators of vesicular transport along exocytic, endocytic, and recycling pathways [334,335][266][267].9. Effect of HTyr and OLE in Gut Microbiota–Brain Axis
9.1. Gut Microbiota
Humans are defined as super-organisms or holobionts that live in harmony with their symbiotic roommates represented by more than 100 trillion microorganisms whose coordinated actions are thought to be important to human life [336][268]. Altogether they form anatomical, physiological, immunological, or evolutionary units. In particular, the human gut harbors a bacterial ecosystem of 1013–1014 bacterial cells and it is also populated by viruses, fungi, and protozoa that collectively form a complex microbial community known as the gut microbiota [337][269]. All these microorganisms are not identified as pathogens by our immune system but, on the contrary, most of them coexist symbiotically with the enterocytes [338][270]. The presence of an enormous number of microbes, which far exceeds the number of human cells, leads to the assumption that the microbiota can influence the physiology of the host organism. This is even more reasonable if wpeople consider that the microbial genome (microbiome) vastly exceeds the human host genome’s size [339][271]. This element alone is sufficient to realize that our symbiotic bacteria are essential for numerous physiological processes that guarantee well-being by ensuring our body’s homeostasis. An imbalanced composition of bacterial populations can be harmful to human health, contributing to the onset of numerous pathologies. However, although most alterations of the gut microbiota turn into an outbreak of diseases and pathologies, actually, a modification in the composition of the gut microbiota is a physiological event. It has been reported that the gut microbiota plays critical roles in the maintenance of human health: (i) taking part in the digestion of food substances, facilitating access to nutrients that would otherwise be inaccessible to the host; (ii) promoting host cells differentiation to protect them from pathogens; (iii) stimulating and modulating the immune system [340][272]. The human intestine is composed of a balanced microbiota with two dominant phyla accounting for about 90% of the total, i.e., Bacteroidetes and Firmicutes, and four less represented phyla, such as Proteobacteria, Actinobacteria, Fusobacteria, and Verrucomicrobia [341][273]. Such microorganisms can be autochthonous (indigenous) or allochthonous (transient) and are symbionts in most cases. Despite this, they are considered pathogenic when assuming opportunistic behavior to the detriment of the host [337][269]. The Firmicutes/Bacteroidetes ratio is an important parameter for emphasizing a potential gut microbiota disorder [342,343,344,345][274][275][276][277] but the abundance, diversity, and homogeneity of the intestinal microbiota are also indicators of a state of health. Nevertheless, this ratio can also be linked to physiological changes in bacterial profiles during different stages of life. Indeed, it is low in the first years of life, then increases in adulthood and decreases again in old age [346][278]. Overall, the composition of the microbiota is unique to each individual because it can be influenced by different factors acting throughout life [347][279]. Indeed, there are many factors such as genetics, diet, environment, exposure to drugs or more generally lifestyle, which influence the composition of the microbiota allowing the proliferation of certain species, rather than others. This aspect is extremely important as the conditions that favor the balanced assembly of microbial populations beneficial to the host determine the establishment of homeostasis defined as “eubiosis”. If, on the other hand, factors come into play that destabilize this condition of equilibrium, the so-called state of “dysbiosis” occurs. Changing the composition and function of the gut microbiota can alter intestinal permeability, digestion, and metabolism as well as immune responses. In particular, an altered state of the gut microbiota causes a pro-inflammatory state, and this condition can lead to the onset of many diseases ranging from gastrointestinal and metabolic conditions to immunological and neuropsychiatric diseases [348][280].9.2. Gut Microbiota across the Lifespan
The intestinal microbiota establishes a co-evolution relationship with the host (which in fact represents its ecosystem) and its development is regulated by a complex interaction between the host and environmental factors, such as diet and lifestyle. For this reason, knowing the transformation of the gut microbiota from birth to old age may shed light on the variation of this community during lifespan and on the possible associations with disease risks. The symbiosis with the microbiota is established from birth and is rewired several times in the first years of life, a period during which children undergo rapid and irreversible growth, showing significant increases in height and weight, and their organs and cognitive abilities undergo great changes [349][281]. Numerous scientific papers reported that the establishment of this symbiosis in childhood is of the utmost importance and that imbalances in intestinal microbiota composition during infancy are associated with various metabolic, immune, and neurological diseases. Thus, early childhood offers a unique opportunity to modulate the gut microbiota in order to promote long-term health [349][281]. At first, the intestinal microbial flora of the newborn shows a low biodiversity, which however will increase during development. During the first days of life, Proteobacteria and Firmicutes represent the two most abundant phyla in vaginally born infants; while from the 7th to the 15th day after birth, Actinobacteria appeared in the feces of cesarean-delivered infants [350][282]. Around the age of three, the intestinal microbiota will move from a highly unstable and poorly differentiated composition to a more stable composition with the typical characteristics of the adult intestinal microbiota [351][283]. From this point on, the gut microbiota will rest in a stable state from the third to the seventh decade of life, although the proportions of Bifidobacteria, Firmicutes, and Fecalibacterium prausnitzii tend to decrease with an increase in Escherichia coli, Proteobacteria, and Staphylococcus [338,352][270][284]. Research carried out on the dynamics of the microbiota in elderly individuals has also provided further information on the possible trajectories of the intestinal microflora throughout human life [353][285]. While the composition of the adult human gut microbiota is generally stable if unperturbed, its stability deteriorates in old age [352][284] and alterations causing dysbiosis are becoming more and more frequent due to age-related factors. For example, a decrease in Bifidobacterium and an increase in Clostridium and Proteobacteria have been observed in older people [338][270]. Given the role of Bifidobacterium in stimulating the immune system and metabolic processes, its decrease could partly explain the compromised immune system in the elderly [341][273]. Furthermore, the greater possibility of developing dysbiosis in old age is a factor to be correlated to the onset of neurodegenerative diseases.9.3. Gut Microbiota–Brain Axis
The trade-off between gut microbiota and brain is now regarded as a pivotal hub for healthy life and aging. The so-called microbiota–gut–brain axis is considered a neuroendocrine system and implies a bidirectional communication between the gut microbiota and the brain, whose dysregulation has emerged to affect host health and disease [354][286]. A healthy host–microbial balance is fundamental to maintain the physical and mental health of both young and elderly populations, while dysbiosis is steadily more implicated in the onset of metabolic, inflammatory, and neurological disorders [347][279]. It is now clear that alterations in top-down (brain to gut) communications are associated with gut inflammation syndromes and appetite disturbances, whereas dysregulations in the bottom-up (gut to brain) interactions are associated with alterations in nervous system functions and neurologic pathologies [355][287]. The relationship between microbiota and brain is known to be regulated at multiple levels, such as immunological (cytokines), endocrine (cortisol), and neuronal, including both central (CNS) and enteric (ENS) nervous systems interconnected by the vagus nerve [354][286]. The short-chain fatty acids (SCFAs), formed by microbial processing of dietary indigestible fibers, are emerging as key players in neuro-immunoendocrine regulation. Indeed, SCFAs can be used locally by colonocytes as an energy source but are also effective in the maintenance of host intestinal barrier integrity and immunity, suppressing cytokines production by myeloid cells and inducing regulatory T-cell differentiation [356][288]. At the colon level, SCFAs also induce the secretion of anorexigenic peptides, which act on hypothalamic centers regulating nutritional habits and energy balance [357][289]. In addition to local effects, SCFAs can enter the bloodstream to be distributed to other organs and, after crossing the blood–brain barrier (BBB), contribute to its integrity, inducing the expression of tight junction proteins, and can modulate brain and behavior [358][290]). Moreover, in the CNS, SCFAs influence neuroinflammation by regulating the maturation and function of microglia as well as by modulating the levels of neurotrophic factors, increasing neurogenesis, contributing to the biosynthesis of serotonin, and improving neuronal homeostasis and function [356][288]. The gut microbiota may influence the functions of ENS and CNS nervous systems also by producing metabolites and neurotransmitters with neuromodulatory properties, such as gamma-aminobutyric acid (GABA), noradrenaline, dopamine, serotonin (5-HT), and their precursors (e.g., tryptophan and tyrosine). In turn, the host nervous system modulates the motility of the gastrointestinal (GI) tract and the intestinal barrier homeostasis, sustaining the microbial community [355][287]. Studies usin g germ-free (GF) mice (i.e., axenic, free of all microorganisms) or antibiotic-treated specific pathogen-free (SPF) mice (i.e., free of a specific list of pathogens) have provided the strongest proof of the importance of microbiota in gut–brain signaling. GF mice have been demonstrated to have altered behavior, impaired immune systems, dysregulated hormone signaling, abnormal metabolism and neurotransmission with respect to their standard counterparts [359][291]. For example, GF mice were reported to show hyperactivity of the hypothalamic–pituitary–adrenal (HPA) axis upon restraint stress, which could be reversed by administration of Bifidobacterium infantis, but not by monocolonization with the enteropathogenic bacteria E. coli [360[292][293],361], highlighting the influence of microbiota on stress responsivity. In addition, a complete SPF flora was able to partly reverse the HPA response to stress only when it was introduced at an early stage of development, suggesting that brain sensitivity to gut signals may occur only within a critical time window [360][292]. Anxiety-like behaviors were also found to be affected in GF mice, which frequently showed an increased exploratory and locomotor behavior, as an index of reduced anxiety [362,363,364][294][295][296]. Furthermore, altered expression of synaptic plasticity-related genes, including BDNF and nerve growth factor-inducible clone A (NGFI-A) [364[296][297],365], as well as variable plasma levels of neurotransmitters [363][295], have been reported in GF mice. However, it is worth noting that the results of the studies on GF mice may vary depending on age, gender, and strains used, as reviewed by Cryan et al. (2019) [347][279]. Besides being an important risk factor for the development of many diseases, ranging from cardiovascular complications to neurologic illnesses, gut dysbiosis has been also demonstrated to affect post-disease recovery. For example, emerging experimental and clinical evidence showed the influence of gut microbiota not only on ischemic stroke pathogenesis but also on treatment outcomes. In fact, ischemic brain dysregulates intestinal homeostasis directing aberrated signals to the intestine either via the neural or HPA axis pathways, resulting in poor stroke treatment prognosis [366][298]. On the other hand, the transplantation of gut microbiota from normal mice into the intestinal tract of mice with ischemic stroke improved the long-term prognosis and survival rate [367][299]. Therefore, therapeutic approaches targeting gut dysbiosis can be considered as promising tools for the treatment and management of stroke or, more in general, of various age-related disorders in which gut dysbiosis may have a role, including neurodegenerative, cardiovascular, metabolic, and musculoskeletal diseases, as well as immune system diseases and cancer [368,369,370,371][300][301][302][303].9.4. Effects of HTyr and OLE on Gut Microbiota under Pathological Conditions
It is now well-established that dietary habits represent one of the main factors determining the composition of the gut microbiota. The Mediterranean diet, which is rich in foods of plant origin, is universally recognized as healthy, and it is amply demonstrated that EVOO, its main source of fat, brings benefits to human health influencing the gut microbiota composition [372][304]. In general, ingested polyphenols have been reported to counteract oxidative stress and inflammatory injury, thus protecting the digestive system from chronic and recurrent diseases. Furthermore, it is widely recognized that they are able to exert neuroprotection and promote cognitive functions, but their connection with the gut microbiota is a more recent discovery. Several studies reported the beneficial effect of EVOO or, more in general of polyphenols, on intestinal microbiota [72[49][304][305][306],372,373,374], whereas less is known about the effect of the exclusive administration of HTyr and OLE. Thus, in this review, we wsection will focus on the outcome of HTyr and OLE on gut microbiota shaping and its implications on healthy host aging. Unlike HTyr and tyrosol, which are well absorbed in the small intestine, OLE reaches the colon unchanged and is rapidly degraded by the colonic microflora to produce HTyr, which can then be absorbed [375][307]. OLE is preferentially degraded in vivo by lactic acid bacteria, such as Lactobacillus and Bifidobacterium species [376][308], which use OLE as a carbon source and benefit from its metabolism. HTyr administration in high-fat-diet-fed mice was found to increase the concentration of the probiotic bacteria Lactobacillus, specifically L. johnsonii, and to not significantly change the ratio Firmicutes/Bacteroidetes (F/B), which was instead decreased in normal-diet-fed mice, indicating that HTyr may be beneficial for metabolism and may control F/B ratio [377][309]. Dietary HTyr supplementation in high-fat-diet-fed mice also reduced the numbers of Proteobacteria and Deferribacteres, whose abundances have been associated with dysbiosis in hosts with metabolic or inflammatory disorders [378][310], and promoted intestinal integrity reducing inflammation level [377][309]. The ability of HTyr to stabilize the F/B ratio and restrain the increase of Proteobacteria may also help controlling gut dysbiosis in inflammatory bowel disease patients, in which a decrease of Firmicutes and Bacteroidetes and an increase of Proteobacteria are the most consistent hallmarks of disease [379][311]. The beneficial effect of HTyr and OLE on gut microbiota is also boosted by their ability to inhibit pathogenic bacteria growth, either Gram-positive or Gram-negative, such as E. coli [72,380][49][312]. The HTyr-induced modulation of colon microbiota was also correlated to its powerful antioxidant effect in oxidative-stressed mice [178][120]. In fact, the HTyr-promoted decrease of the lipid peroxidation marker malondialdehyde (MDA) in the serum was correlated with the increase of the relative abundance of Firmicutes and Lactobacillus and the decrease of Bacteroidetes, as an effect of HTyr administration [178][120]. Moreover, HTyr decreased Parabacteroides, whose abundance was associated with the progression of oxidative stress and inflammation [178][120]. Interestingly, the HTyr-induced increase of Firmicutes positively correlated with a higher concentration of butyrate, one of the most important SCFAs, involved not only in maintaining intestinal function but also in reducing oxidative stress [381][313]). Surprisingly, HTyr supplementation was demonstrated to be effective in relieving oxidative stress in mice also by increasing Staphylococcales [382][314], whose success as pathogens is due in part to their ability to mitigate endogenous and exogenous oxidative and nitrosative stress [383][315]. The restrain of oxidative stress due to HTyr administration was also found to prevent fine dust (PM2.5)-induced adiposity and insulin resistance in adult mice [384][316]. Once again, it was highlighted that gut microbiota may mediate the actions of HTyr, which was effective in increasing microbiota richness, reducing pathogenic bacteria, and reversing PM2.5-induced gut dysbiosis. In particular, the genus Akkermansia, belonging to the Verrucomicrobia phylum and known to be beneficial for reducing weight gain and endotoxemia in mice and humans [385][317], was exclusively abundant in HTyr-administered mice. In addition, Ruminococcaceae and Mycoplasmataceae, beneficial for glucose metabolism and abundant in long-living people, were enhanced by HTyr administration as well as Prevotellaceae, which negatively correlated with lipid peroxidation biomarkers, thus contributing to the HTyr antioxidant effect [384][316]. Besides HTyr, OLE was also found to ameliorate advanced-stage type 2 diabetes in mice by regulating gut microbiota [386][318]. Consistent with the previously reported effects of HTyr administration, O LE was shown to significantly reduce the relative abundance of Bacteroidetes, including the Parabacteroides genus, and increase that of Verrucomicrobia, including the Akkermansia genus, confirming their importance in improving diabetes symptoms and insulin resistance [386][318]. Altogether, the above studies highlighted that HTyr and OLE both act as prebiotics in gut microbiota modulation, favoring intestinal microbes’ homeostasis, even in the presence of chronic diseases. Their administration has been demonstrated to increase the diversity of gut microbiota, enhancing beneficial bacteria, and inhibiting pathogenic ones, thus impacting the health of multiple organs and systems. In fact, the well-established relationship between gut and CNS highlights the role of prebiotics in reducing the degree of oxidative stress and consequent progression of neurodegenerative and cardiovascular diseases and metabolic syndromes. For this reason, the maintenance of a condition of eubiosis is crucial to guarantee healthy aging, preventing and ameliorating several chronic pathologies. The potential therapeutic application of prebiotics supplied with the diet is increasingly investigated in order to keep optimal health during aging and also for the treatment of age-related disorders.References
- Lopez de las Hazas, M.-C.L.D.L.; Piñol, C.; Macià, A.; Romero, M.-P.; Pedret, A.; Solà, R.; Rubió, L.; Motilva, M.-J. Differential absorption and metabolism of hydroxytyrosol and its precursors oleuropein and secoiridoids. J. Funct. Foods 2016, 22, 52–63.
- Slavin, J.L.; Lloyd, B. Health Benefits of Fruits and Vegetables. Adv. Nutr. Int. Rev. J. 2012, 3, 506–516.
- Liu, R.H. Health-Promoting Components of Fruits and Vegetables in the Diet. Adv. Nutr. Int. Rev. J. 2013, 4, 384S–392S.
- Roleira, F.M.F.; Tavares-Da-Silva, E.J.; Varela, C.L.; Costa, S.C.; Silva, T.; Garrido, J.; Borges, F. Plant derived and dietary phenolic antioxidants: Anticancer properties. Food Chem. 2015, 183, 235–258.
- Aune, D.; Giovannucci, E.; Boffetta, P.; Fadnes, L.T.; Keum, N.N.; Norat, T.; Greenwood, D.C.; Riboli, E.; Vatten, L.J.; Tonstad, S. Fruit and vegetable intake and the risk of cardiovascular disease, total cancer and all-cause mortality—A systematic review and dose-response meta-analysis of prospective studies. Int. J. Epidemiol. 2017, 46, 1029–1056.
- Borek, C. Aging and antioxidants. Fruits and vegetables are powerful armor. Adv. Nurse Pract. 2006, 14, 35–38.
- Bach-Faig, A.; Berry, E.M.; Lairon, D.; Reguant, J.; Trichopoulou, A.; Dernini, S.; Medina, F.X.; Battino, M.; Belahsen, R.; Miranda, G.; et al. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011, 14, 2274–2284.
- Schwingshackl, L.; Morze, J.; Hoffmann, G. Mediterranean diet and health status: Active ingredients and pharmacological mechanisms. Br. J. Pharmacol. 2020, 177, 1241–1257.
- Hu, T.; He, X.-W.; Jiang, J.-G.; Xu, X.-L. Hydroxytyrosol and Its Potential Therapeutic Effects. J. Agric. Food Chem. 2014, 62, 1449–1455.
- Visioli, F.; Davalos, A.; Hazas, M.D.C.L.D.L.; Crespo, M.D.C.; Tomé-Carneiro, J. An overview of the pharmacology of olive oil and its active ingredients. Br. J. Pharmacol. 2020, 177, 1316–1330.
- de Bock, M.; Thorstensen, E.B.; Derraik, J.G.B.; Henderson, H.V.; Hofman, P.L.; Cutfield, W.S. Human absorption and metabolism of oleuropein and hydroxytyrosol ingested as olive (Olea europaea L.) leaf extract. Mol. Nutr. Food Res. 2013, 57, 2079–2085.
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379.
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Food Systems: A Mini-Review. Front. Nutr. 2018, 5, 87.
- Brigelius-Flohé, R. Vitamin E research: Past, now and future. Free. Radic. Biol. Med. 2021, 177, 381–390.
- Lombardo, L.; Grasso, F.; Lanciano, F.; Loria, S.; Monetti, E. Broad-Spectrum Health Protection of Extra Virgin Olive Oil Compounds. Stud. Nat. Prod. 2018, 57, 41–77.
- Karković Marković, A.; Torić, J.; Barbarić, M.; Jakobusić Brala, C. Hydroxytyrosol, Tyrosol and Derivatives and Their Potential Effects on Human Health. Molecules 2019, 24, 2001.
- Gambacorta, A.; Tofani, D.; Bernini, R.; Migliorini, A. High-Yielding Preparation of a Stable Precursor of Hydroxytyrosol by Total Synthesis and from the Natural Glycoside Oleuropein. J. Agric. Food Chem. 2007, 55, 3386–3391.
- Lo Scalzo, R.; Scarpati, M.L.; Verzegnassi, B.; Vita, G. Olea europaea chemicals repellent toDacus oleae females. J. Chem. Ecol. 1994, 20, 1813–1823.
- Jemai, H.; El Feki, A.; Sayadi, S. Antidiabetic and Antioxidant Effects of Hydroxytyrosol and Oleuropein from Olive Leaves in Alloxan-Diabetic Rats. J. Agric. Food Chem. 2009, 57, 8798–8804.
- Romani, A.; Ieri, F.; Urciuoli, S.; Noce, A.; Marrone, G.; Nediani, C.; Bernini, R. Health Effects of Phenolic Compounds Found in Extra-Virgin Olive Oil, By-Products, and Leaf of Olea europaea L. Nutrients 2019, 11, 1776.
- Rietjens, S.J.; Bast, A.; Haenen, G.R.M.M. New Insights into Controversies on the Antioxidant Potential of the Olive Oil Antioxidant Hydroxytyrosol. J. Agric. Food Chem. 2007, 55, 7609–7614.
- Bernini, R.; Merendino, N.; Romani, A.; Velotti, F. Naturally Occurring Hydroxytyrosol: Synthesis and Anticancer Potential. Curr. Med. Chem. 2013, 20, 655–670.
- Vilaplana-Perez, C.; Aunon, D.; Garcia-Flores, L.A.; Gil-Izquierdo, A. Hydroxytyrosol and Potential Uses in Cardiovascular Diseases, Cancer, and AIDS. Front. Nutr. 2014, 1, 18.
- D’Andrea, G.; Ceccarelli, M.; Bernini, R.; Clemente, M.; Santi, L.; Caruso, C.; Micheli, L.; Tirone, F. Hydroxytyrosol stimulates neurogenesis in aged dentate gyrus by enhancing stem and progenitor cell proliferation and neuron survival. FASEB J. 2020, 34, 4512–4526.
- Franconi, F.; Campesi, I.; Romani, A. Is Extra Virgin Olive Oil an Ally for Women’s and Men’s Cardiovascular Health? Cardiovasc. Ther. 2020, 2020, 6719301.
- Pizzichini, M.; Russo, C. Process for Recovering the Components of Olive Mill Wastewater with Membrane Technologies. Patent WO 2005123603A1, 29 December 2005.
- Sabatini, N. Recent Patents in Olive Oil Industry: New Technologies for the Recovery of Phenols Compounds from Olive Oil, Olive Oil Industrial By-Products and Waste Waters. Recent Patents Food Nutr. Agric. 2010, 2, 154–159.
- Stahel, W.R. The circular economy. Nature 2016, 531, 435–438.
- Capasso, R.; Evidente, A.; Avolio, S.; Solla, F. A Highly Convenient Synthesis of Hydroxytyrosol and Its Recovery from Agricultural Waste Waters. J. Agric. Food Chem. 1999, 47, 1745–1748.
- Espín, J.C.; Soler-Rivas, C.; Cantos, E.; Tomás-Barberán, F.A.; Wichers, H.J. Synthesis of the Antioxidant Hydroxytyrosol Using Tyrosinase as Biocatalyst. J. Agric. Food Chem. 2001, 49, 1187–1193.
- Bernini, R.; Mincione, E.; Barontini, M.; Crisante, F. Convenient Synthesis of Hydroxytyrosol and Its Lipophilic Derivatives from Tyrosol or Homovanillyl Alcohol. J. Agric. Food Chem. 2008, 56, 8897–8904.
- Guazzaroni, M.; Crestini, C.; Saladino, R. Layer-by-Layer coated tyrosinase: An efficient and selective synthesis of catechols. Bioorganic Med. Chem. 2012, 20, 157–166.
- Bonacci, S.; Paonessa, R.; Costanzo, P.; Salerno, R.; Maiuolo, J.; Nardi, M.; Procopio, A.; Manuela, O. Peracetylation as a strategy to improve oleuropein stability and its affinity to fatty foods. Food Funct. 2018, 9, 5759–5767.
- Grasso, S.; Siracusa, L.; Spatafora, C.; Renis, M.; Tringali, C. Hydroxytyrosol lipophilic analogues: Enzymatic synthesis, radical scavenging activity and DNA oxidative damage protection. Bioorganic Chem. 2007, 35, 137–152.
- Bouallagui, Z.; Bouaziz, M.; Lassoued, S.; Engasser, J.M.; Ghoul, M.; Sayadi, S. Hydroxytyrosol Acyl Esters: Biosynthesis and Activities. Appl. Biochem. Biotechnol. 2010, 163, 592–599.
- Calderón-Montaño, J.M.; Madrona, A.; Burgos-Morón, E.; Orta, M.L.; Mateos, S.; Espartero, J.L.; López-Lázaro, M. Selective Cytotoxic Activity of New Lipophilic Hydroxytyrosol Alkyl Ether Derivatives. J. Agric. Food Chem. 2013, 61, 5046–5053.
- Bernini, R.; Gilardini Montani, M.S.; Merendino, N.; Romani, A.; Velotti, F. Hydroxytyrosol-Derived Compounds: A Basis for the Creation of New Pharmacological Agents for Cancer Prevention and Therapy. J. Med. Chem. 2015, 58, 9089–9107.
- Bernini, R.; Carastro, I.; Santoni, F.; Clemente, M. Synthesis of Lipophilic Esters of Tyrosol, Homovanillyl Alcohol and Hydroxytyrosol. Antioxidants 2019, 8, 174.
- Bernini, R.; Crisante, F.; Merendino, N.; Molinari, R.; Soldatelli, M.C.; Velotti, F. Synthesis of a novel ester of hydroxytyrosol and α-lipoic acid exhibiting an antiproliferative effect on human colon cancer HT-29 cells. Eur. J. Med. Chem. 2011, 46, 439–446.
- Martin, D.; Moran-Valero, M.I.; Casado, V.; Reglero, G.; Torres, C.F. Phosphatidyl Derivative of Hydroxytyrosol. In Vitro Intestinal Digestion, Bioaccessibility, and Its Effect on Antioxidant Activity. J. Agric. Food Chem. 2014, 62, 9751–9759.
- Roma, E.; Mattoni, E.; Lupattelli, P.; Moeini, S.; Gasperi, T.; Bernini, R.; Incerpi, S.; Tofani, D. New Dihydroxytyrosyl Esters from Dicarboxylic Acids: Synthesis and Evaluation of the Antioxidant Activity In Vitro (ABTS) and in Cell-Cultures (DCF Assay). Molecules 2020, 25, 3135.
- Romanucci, V.; Giordano, M.; de Tommaso, G.; Iuliano, M.; Bernini, R.; Clemente, M.; Garcia-Viñuales, S.; Milardi, D.; Zarrelli, A.; Di Fabio, G. Synthesis of New Tyrosol-Based Phosphodiester Derivatives: Effect on Amyloid β Aggregation and Metal Chelation Ability. Chemmedchem 2021, 16, 1172–1183.
- Bonechi, C.; Donati, A.; Tamasi, G.; Pardini, A.; Rostom, H.; Leone, G.; Lamponi, S.; Consumi, M.; Magnani, A.; Rossi, C. Chemical characterization of liposomes containing nutraceutical compounds: Tyrosol, hydroxytyrosol and oleuropein. Biophys. Chem. 2019, 246, 25–34.
- De Luca, I.; di Cristo, F.; Valentino, A.; Peluso, G.; di Salle, A.; Calarco, A. Food-Derived Bioactive Molecules from Mediterranean Diet: Nanotechnological Approaches and Waste Valorization as Strategies to Improve Human Wellness. Polymers 2022, 14, 1726.
- Monteiro, M.; Silva, A.F.R.; Resende, D.; Braga, S.S.; Coimbra, M.A.; Silva, A.M.S.; Cardoso, S.M. Strategies to Broaden the Applications of Olive Biophenols Oleuropein and Hydroxytyrosol in Food Products. Antioxidants 2021, 3, 444.
- Vissers, M.N.; Zock, P.L.; Roodenburg, A.J.C.; Leenen, R.; Katan, M.B. Olive Oil Phenols Are Absorbed in Humans. J. Nutr. 2002, 132, 409–417.
- Miro-Casas, E.; Covas, M.-I.; Farre, M.; Fito, M.; Ortuño, J.; Weinbrenner, T.; Roset, P.; de la Torre, R. Hydroxytyrosol Disposition in Humans. Clin. Chem. 2003, 49, 945–952.
- Edgecombe, S.C.; Stretch, G.L.; Hayball, P.J. Oleuropein, an Antioxidant Polyphenol from Olive Oil, Is Poorly Absorbed from Isolated Perfused Rat Intestine. J. Nutr. 2000, 130, 2996–3002.
- Deiana, M.; Serra, G.; Corona, G. Modulation of intestinal epithelium homeostasis by extra virgin olive oil phenolic compounds. Food Funct. 2018, 9, 4085–4099.
- Rodríguez-López, P.; Lozano-Sanchez, J.; Borrás-Linares, I.; Emanuelli, T.; Menéndez, J.; Segura-Carretero, A. Structure–Biological Activity Relationships of Extra-Virgin Olive Oil Phenolic Compounds: Health Properties and Bioavailability. Antioxidants 2020, 9, 685.
- Roowi, S.; Mullen, W.; Edwards, C.A.; Crozier, A. Yoghurt impacts on the excretion of phenolic acids derived from colonic breakdown of orange juice flavanones in humans. Mol. Nutr. Food Res. 2009, 53 (Suppl. 1), S68–S75.
- Visioli, F.; Galli, C.; Grande, S.; Colonnelli, K.; Patelli, C.; Galli, G.; Caruso, D. Hydroxytyrosol Excretion Differs between Rats and Humans and Depends on the Vehicle of Administration. J. Nutr. 2003, 133, 2612–2615.
- Tuck, K.L.; Freeman, M.P.; Hayball, P.J.; Stretch, G.L.; Stupans, I. The In Vivo Fate of Hydroxytyrosol and Tyrosol, Antioxidant Phenolic Constituents of Olive Oil, after Intravenous and Oral Dosing of Labeled Compounds to Rats. J. Nutr. 2001, 131, 1993–1996.
- Malliou, F.; Andriopoulou, C.E.; Gonzalez, F.J.; Kofinas, A.; Skaltsounis, A.-L.; Konstandi, M. Oleuropein-Induced Acceleration of Cytochrome P450–Catalyzed Drug Metabolism: Central Role for Nuclear Receptor Peroxisome Proliferator-Activated Receptor α. Drug Metab. Dispos. 2021, 49, 833–843.
- Zhou, S.; Chan, E.; Pan, S.-Q.; Huang, M.; Lee, E.J.D. Pharmacokinetic Interactions of Drugs with St John’s Wort. J. Psychopharmacol. 2004, 18, 262–276.
- Sakavitsi, M.E.; Breynaert, A.; Nikou, T.; Lauwers, S.; Pieters, L.; Hermans, N.; Halabalaki, M. Availability and Metabolic Fate of Olive Phenolic Alcohols Hydroxytyrosol and Tyrosol in the Human GI Tract Simulated by the In Vitro GIDM–Colon Model. Metabolites 2022, 12, 391.
- Bai, C.; Yan, X.; Takenaka, M.; Sekiya, K.; Nagata, T. Determination of Synthetic Hydroxytyrosol in Rat Plasma by GC-MS. J. Agric. Food Chem. 1998, 46, 3998–4001.
- Domínguez-Perles, R.; Auñón, D.; Ferreres, F.; Gil-Izquierdo, A. Gender differences in plasma and urine metabolites from Sprague–Dawley rats after oral administration of normal and high doses of hydroxytyrosol, hydroxytyrosol acetate, and DOPAC. Eur. J. Nutr. 2015, 56, 215–224.
- Fernández-Ávila, C.; Montes, R.; Castellote, A.I.; Chisaguano, A.M.; Fitó, M.; Covas, M.I.; Muñoz-Aguallo, D.; Nyyssönen, K.; Zunft, H.J.; López-Sabater, M.C. Fast determination of virgin olive oil phenolic metabolites in human high-density lipoproteins. Biomed. Chromatogr. 2015, 29, 1035–1041.
- D’Angelo, S.; Manna, C.; Migliardi, V.; Mazzoni, O.; Morrica, P.; Capasso, G.; Pontoni, G.; Galletti, P.; Zappia, V. Pharmacokinetics and metabolism of hydroxytyrosol, a natural antioxidant from olive oil. Drug Metab. Dispos. 2001, 29, 1492–1498.
- Granados-Principal, S.; El-Azem, N.; Pamplona, R.; Ramírez-Tortosa, C.; Pulido-Morán, M.; Ramirez, L.V.; Quiles, J.L.; Sanchez-Rovira, P.; Naudí, A.; Portero-Otin, M.; et al. Hydroxytyrosol ameliorates oxidative stress and mitochondrial dysfunction in doxorubicin-induced cardiotoxicity in rats with breast cancer. Biochem. Pharmacol. 2014, 90, 25–33.
- López de las Hazas, M.C.; Rubió, L.; Kotronoulas, A.; de la Torre, R.; Solà, R.; Motilva, M.J. Dose effect on the uptake and accumulation of hydroxytyrosol and its metabolites in target tissues in rats. Mol. Nutr. Food Res. 2015, 59, 1395–1399.
- Weinbrenner, T.; Fitó, M.; de la Torre, R.; Saez, G.T.; Rijken, P.; Tormos, C.; Coolen, S.; Albaladejo, M.F.; Abanades, S.; Schroder, H.; et al. Olive Oils High in Phenolic Compounds Modulate Oxidative/Antioxidative Status in Men. J. Nutr. 2004, 134, 2314–2321.
- Khymenets, O.; Farré, M.; Pujadas, M.; Ortiz, E.; Joglar, J.; Covas, M.I.; de la Torre, R. Direct analysis of glucuronidated metabolites of main olive oil phenols in human urine after dietary consumption of virgin olive oil. Food Chem. 2011, 126, 306–314.
- Rodríguez-Morató, J.; Boronat, A.; Kotronoulas, A.; Pujadas, M.; Pastor, A.; Olesti, E.; Pérez-Mañá, C.; Khymenets, O.; Fitó, M.; Farré, M.; et al. Metabolic disposition and biological significance of simple phenols of dietary origin: Hydroxytyrosol and tyrosol. Drug Metab. Rev. 2016, 48, 218–236.
- Serreli, G.; Deiana, M. Biological Relevance of Extra Virgin Olive Oil Polyphenols Metabolites. Antioxidants 2018, 7, 170.
- de la Torre, R.; Corella, D.; Castañer, O.; A Martínez-González, M.; Salas-Salvadó, J.; Vila-Domènech, J.S.; Estruch, R.; Sorli, J.V.; Arós, F.; Fiol, M.; et al. Protective effect of homovanillyl alcohol on cardiovascular disease and total mortality: Virgin olive oil, wine, and catechol-methylathion. Am. J. Clin. Nutr. 2017, 105, 1297–1340.
- Kotronoulas, A.; Pizarro, N.; Serra, A.; Robledo, P.; Joglar, J.; Rubió, L.; Hernáez, Á.; Tormos, C.; Motilva, M.J.; Fitó, M.; et al. Dose-dependent metabolic disposition of hydroxytyrosol and formation of mercapturates in rats. Pharmacol. Res. 2013, 77, 47–56.
- Robles-Almazan, M.; Pulido-Moran, M.; Moreno-Fernandez, J.; Ramirez-Tortosa, C.; Rodriguez-Garcia, C.; Quiles, J.L.; Ramirez-Tortosa, M. Hydroxytyrosol: Bioavailability, toxicity, and clinical applications. Food Res. Int. 2018, 105, 654–667.
- Suárez, M.; Valls, R.M.; Romero, M.-P.; Macià, A.; Fernández, S.; Giralt, M.; Solà, R.; Motilva, M.-J. Bioavailability of phenols from a phenol-enriched olive oil. Br. J. Nutr. 2011, 106, 1691–1701.
- Galmés, S.; Reynés, B.; Palou, M.; Palou-March, A.; Palou, A. Absorption, Distribution, Metabolism, and Excretion of the Main Olive Tree Phenols and Polyphenols: A Literature Review. J. Agric. Food Chem. 2021, 69, 5281–5296.
- Chashmi, N.A.; Emadi, S.; Khastar, H. Protective effects of hydroxytyrosol on gentamicin induced nephrotoxicity in mice. Biochem. Biophys. Res. Commun. 2017, 482, 1427–1429.
- Kano, S.; Komada, H.; Yonekura, L.; Sato, A.; Nishiwaki, H.; Tamura, H. Absorption, Metabolism, and Excretion by Freely Moving Rats of 3,4-DHPEA-EDA and Related Polyphenols from Olive Fruits (Olea europaea). J. Nutr. Metab. 2016, 2016, 1–10.
- Aunon-Calles, D.; Canut, L.; Visioli, F. Toxicological evaluation of pure hydroxytyrosol. Food Chem. Toxicol. 2013, 55, 498–504.
- Christian, M.S.; Sharper, V.A.; Hoberman, A.M.; Seng, J.E.; Fu, L.; Covell, D.; Diener, R.M.; Bitler, C.M.; Crea, R. The Toxicity Profile of Hydrolyzed Aqueous Olive Pulp Extract. Drug Chem. Toxicol. 2004, 27, 309–330.
- Lee, Y.Y.; Crauste, C.; Wang, H.; Leung, H.H.; Vercauteren, J.; Galano, J.-M.; Oger, C.; Durand, T.; Wan, J.M.-F.; Lee, J.C.-Y. Extra Virgin Olive Oil Reduced Polyunsaturated Fatty Acid and Cholesterol Oxidation in Rodent Liver: Is This Accounted for Hydroxytyrosol-Fatty Acid Conjugation? Chem. Res. Toxicol. 2016, 29, 1689–1698.
- Bassani, B.; Rossi, T.; de Stefano, D.; Pizzichini, D.; Corradino, P.; Macrì, N.; Noonan, D.M.; Albini, A.; Bruno, A. Potential chemopreventive activities of a polyphenol rich purified extract from olive mill wastewater on colon cancer cells. J. Funct. Foods 2016, 27, 236–248.
- Bernini, R.; Carastro, I.; Palmini, G.; Tanini, A.; Zonefrati, R.; Pinelli, P.; Brandi, M.L.; Romani, A. Lipophilization of Hydroxytyrosol-Enriched Fractions from Olea europaea L. Byproducts and Evaluation of the in Vitro Effects on a Model of Colorectal Cancer Cells. J. Agric. Food Chem. 2017, 65, 6506–6512.
- Han, J.; Talorete, T.P.N.; Yamada, P.; Isoda, H. Anti-proliferative and apoptotic effects of oleuropein and hydroxytyrosol on human breast cancer MCF-7 cells. Cytotechnology 2009, 59, 45–53.
- Della Ragione, F.; Cucciolla, V.; Borriello, A.; Della Pietra, V.; Pontoni, G.; Racioppi, L.; Manna, C.; Galletti, P.; Zappia, V. Hydroxytyrosol, a Natural Molecule Occurring in Olive Oil, Induces Cytochrome c-Dependent Apoptosis. Biochem. Biophys. Res. Commun. 2000, 278, 733–739.
- Goldsmith, C.D.; Bond, D.R.; Jankowski, H.; Weidenhofer, J.; Stathopoulos, C.E.; Roach, P.D.; Scarlett, C.J. The Olive Biophenols Oleuropein and Hydroxytyrosol Selectively Reduce Proliferation, Influence the Cell Cycle, and Induce Apoptosis in Pancreatic Cancer Cells. Int. J. Mol. Sci. 2018, 19, 1937.
- Burattini, S.; Salucci, S.; Baldassarri, V.; Accorsi, A.; Piatti, E.; Madrona, A.; Espartero, J.L.; Candiracci, M.; Zappia, G.; Falcieri, E. Anti-apoptotic activity of hydroxytyrosol and hydroxytyrosyl laurate. Food Chem. Toxicol. 2013, 55, 248–256.
- Anter, J.; Tasset, I.; Demyda-Peyrás, S.; Ranchal, I.; Moreno-Millán, M.; Romero-Jimenez, M.; Muntané, J.; de Castro, M.D.L.; Muñoz-Serrano, A.; Alonso-Moraga, Á. Evaluation of potential antigenotoxic, cytotoxic and proapoptotic effects of the olive oil by-product “alperujo”, hydroxytyrosol, tyrosol and verbascoside. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2014, 772, 25–33.
- Laghezza-Masci, V.L.; Bernini, R.; Villanova, N.; Clemente, M.; Cicaloni, V.; Tinti, L.; Salvini, L.; Taddei, A.R.; Tiezzi, A.; Ovidi, E. In Vitro Anti-Proliferative and Apoptotic Effects of Hydroxytyrosyl Oleate on SH-SY5Y Human Neuroblastoma Cells. Int. J. Mol. Sci. 2022, 23, 12348.
- Shamshoum, H.; Vlavcheski, F.; Tsiani, E. Anticancer effects of oleuropein. Biofactors 2017, 43, 517–528.
- Imran, M.; Nadeem, M.; Gilani, S.A.; Khan, S.; Sajid, M.W.; Amir, R.M. Antitumor Perspectives of Oleuropein and Its Metabolite Hy-droxytyrosol: Recent Updates. J Food Sci. 2018, 83, 1781–1791.
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781.
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The Role of Tau in Alzheimer’s Disease and Related Disorders. CNS Neurosci. Ther. 2011, 17, 514–524.
- Gandy, S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J. Clin. Investig. 2005, 115, 1121–1129.
- Daccache, A.; Lion, C.; Sibille, N.; Gerard, M.; Slomianny, C.; Lippens, G.; Cotelle, P. Oleuropein and derivatives from olives as Tau aggregation inhibitors. Neurochem. Int. 2011, 58, 700–707.
- Rigacci, S.; Guidotti, V.; Bucciantini, M.; Nichino, D.; Relini, A.; Berti, A.; Stefani, M. Aβ(1-42) Aggregates into Non-Toxic Amyloid Assemblies in the Presence of the Natural Polyphenol Oleuropein Aglycon. Curr. Alzheimer Res. 2011, 8, 841–852.
- Leri, M.; Natalello, A.; Bruzzone, E.; Stefani, M.; Bucciantini, M. Oleuropein aglycone and hydroxytyrosol interfere differently with toxic Aβ1-42 aggregation. Food Chem. Toxicol. 2019, 129, 1–12.
- Nardiello, P.; Pantano, D.; Lapucci, A.; Stefani, M.; Casamenti, F. Diet Supplementation with Hydroxytyrosol Ameliorates Brain Pathology and Restores Cognitive Functions in a Mouse Model of Amyloid-β Deposition. J. Alzheimer’s Dis. 2018, 63, 1161–1172.
- Luccarini, I.; Grossi, C.; Rigacci, S.; Coppi, E.; Pugliese, A.M.; Pantano, D.; la Marca, G.; Dami, T.E.; Berti, A.; Stefani, M.; et al. Oleuropein aglycone protects against pyroglutamylated-3 amyloid-ß toxicity: Biochemical, epigenetic and functional correlates. Neurobiol. Aging 2015, 36, 648–663.
- Pantano, D.; Luccarini, I.; Nardiello, P.; Servili, M.; Stefani, M.; Casamenti, F. Oleuropein aglycone and polyphenols from olive mill waste water ameliorate cognitive deficits and neuropathology. Br. J. Clin. Pharmacol. 2017, 83, 54–62.
- Arunsundar, M.; Shanmugarajan, T.S.; Ravichandran, V. 3,4-Dihydroxyphenylethanol Attenuates Spatio-Cognitive Deficits in an Alzheimer’s Disease Mouse Model: Modulation of the Molecular Signals in Neuronal Survival-Apoptotic Programs. Neurotox. Res. 2015, 27, 143–155.
- Diomede, L.; Rigacci, S.; Romeo, M.; Stefani, M.; Salmona, M. Oleuropein Aglycone Protects Transgenic C. elegans Strains Expressing Aβ42 by Reducing Plaque Load and Motor Deficit. PLoS ONE 2013, 8, e58893.
- Wu, L.; Velander, P.; Liu, D.; Xu, B. Olive Component Oleuropein Promotes β-Cell Insulin Secretion and Protects β-Cells from Amylin Amyloid-Induced Cytotoxicity. Biochemistry 2017, 56, 5035–5039.
- Palazzi, L.; Bruzzone, E.; Bisello, G.; Leri, M.; Stefani, M.; Bucciantini, M.; de Laureto, P.P. Oleuropein aglycone stabilizes the monomeric α-synuclein and favours the growth of non-toxic aggregates. Sci. Rep. 2018, 8, 8337.
- Gallardo-Fernández, M.; Hornedo-Ortega, R.; Cerezo, A.B.; Troncoso, A.M.; García-Parrilla, M.C. Melatonin, protocatechuic acid and hydroxytyrosol effects on vitagenes system against alpha-synuclein toxicity. Food Chem. Toxicol. 2019, 134, 110817.
- Peng, Y.; Hou, C.; Yang, Z.; Li, C.; Jia, L.; Liu, J.; Tang, Y.; Shi, L.; Li, Y.; Long, J.; et al. Hydroxytyrosol mildly improve cognitive function independent of APP processing in APP/PS1 mice. Mol. Nutr. Food Res. 2016, 60, 2331–2342.
- Qin, C.; Hu, S.; Zhang, S.; Zhao, D.; Wang, Y.; Li, H.; Peng, Y.; Shi, L.; Xu, X.; Wang, C.; et al. Hydroxytyrosol Acetate Improves the Cognitive Function of APP/PS1 Transgenic Mice in ERβ-dependent Manner. Mol. Nutr. Food Res. 2021, 65, e2000797.
- Visioli, F.; Rodríguez-Pérez, M.; Gómez-Torres, Ó.; Pintado-Losa, C.; Burgos-Ramos, E. Hydroxytyrosol improves mitochondrial energetics of a cellular model of Alzheimer’s disease. Nutr. Neurosci. 2022, 25, 990–1000.
- Leri, M.; Bertolini, A.; Stefani, M.; Bucciantini, M. EVOO Polyphenols Relieve Synergistically Autophagy Dysregulation in a Cellular Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7225.
- Stacchiotti, A.; Corsetti, G. Natural Compounds and Autophagy: Allies Against Neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 555409.
- St-Laurent-Thibault, C.; Arseneault, M.; Longpré, F.; Ramassamy, C. Tyrosol and hydroxytyrosol, two main components of olive oil, protect N2a cells against amyloid-β-induced toxicity. Involvement of the NF-κB signaling. Curr. Alzheimer Res. 2011, 8, 543–551.
- Crespo, M.C.; Tomé-Carneiro, J.; Pintado, C.; Dávalos, A.; Visioli, F.; Burgos-Ramos, E. Hydroxytyrosol restores proper insulin signaling in an astrocytic model of Alzheimer’s disease. Biofactors 2017, 43, 540–548.
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909.
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276, 2045–2047.
- Micheli, L.; Creanza, T.M.; Ceccarelli, M.; D’Andrea, G.; Giacovazzo, G.; Ancona, N.; Coccurello, R.; Scardigli, R.; Tirone, F. Transcriptome Analysis in a Mouse Model of Premature Aging of Dentate Gyrus: Rescue of Alpha-Synuclein Deficit by Virus-Driven Expression or by Running Restores the Defective Neurogenesis. Front. Cell Dev. Biol. 2021, 9, 696684.
- Janetzky, B.; Hauck, S.; Youdim, M.B.H.; Riederer, P.; Jellinger, K.; Pantucek, F.; Zöchling, R.; Boissl, K.W.; Reichmann, H. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci. Lett. 1994, 169, 126–128.
- Segura-Aguilar, J.; Paris, I.; Muñoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915.
- Napolitano, A.; Manini, P.; d’Ischia, M. Oxidation chemistry of catecholamines and neuronal degeneration: An update. Curr. Med. Chem. 2011, 18, 1832–1845.
- Funakohi-Tago, M.; Sakata, T.; Fujiwara, S.; Sakakura, A.; Sugai, T.; Tago, K.; Tamura, H. Hydroxytyrosol butyrate inhibits 6-OHDA-induced apoptosis through activation of the Nrf2/HO-1 axis in SH-SY5Y cells. Eur. J. Pharmacol. 2018, 834, 246–256.
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247.
- Saito, R.; Suzuki, T.; Hiramoto, K.; Asami, S.; Naganuma, E.; Suda, H.; Iso, T.; Yamamoto, H.; Morita, M.; Baird, L.; et al. Characterizations of Three Major Cysteine Sensors of Keap1 in Stress Response. Mol. Cell. Biol. 2016, 36, 271–284.
- Peng, S.; Zhang, B.; Yao, J.; Duan, D.; Fang, J. Dual protection of hydroxytyrosol, an olive oil polyphenol, against oxidative damage in PC12 cells. Food Funct. 2015, 6, 2091–2100.
- Zrelli, H.; Kusunoki, M.; Miyazaki, H. Role of Hydroxytyrosol-dependent Regulation of HO-1 Expression in Promoting Wound Healing of Vascular Endothelial Cells via Nrf2 De Novo Synthesis and Stabilization. Phytotherapy Res. 2015, 29, 1011–1018.
- Montoya, T.; Soto, M.A.; Castejón, M.L.; Rosillo, M.Á.; Sánchez-Hidalgo, M.; Begines, P.; Fernández-Bolaños, J.G.; Alarcón-De-La-Lastra, C. Peracetylated hydroxytyrosol, a new hydroxytyrosol derivate, attenuates LPS-induced inflammatory response in murine peritoneal macrophages via regulation of non-canonical inflammasome, Nrf2/HO1 and JAK/STAT signaling pathways. J. Nutr. Biochem. 2018, 57, 110–120.
- Han, H.; Zhong, R.; Zhang, S.; Wang, M.; Wen, X.; Yi, B.; Zhao, Y.; Chen, L.; Zhang, H. Hydroxytyrosol attenuates diquat-induced oxidative stress by activating Nrf2 pathway and modulating colonic microbiota in mice. J. Nutr. Biochem. 2022, 113, 109256.
- Achour, I.; Arel-Dubeau, A.-M.; Renaud, J.; Legrand, M.; Attard, E.; Germain, M.; Martinoli, M.-G. Oleuropein Prevents Neuronal Death, Mitigates Mitochondrial Superoxide Production and Modulates Autophagy in a Dopaminergic Cellular Model. Int. J. Mol. Sci. 2016, 17, 1293.
- Yu, G.; Deng, A.; Tang, W.; Ma, J.; Yuan, C.; Ma, J. Hydroxytyrosol induces phase II detoxifying enzyme expression and effectively protects dopaminergic cells against dopamine- and 6-hydroxydopamine induced cytotoxicity. Neurochem. Int. 2016, 96, 113–120.
- Goldstein, D.S.; Jinsmaa, Y.; Sullivan, P.; Holmes, C.; Kopin, I.J.; Sharabi, Y. 3,4-Dihydroxyphenylethanol (Hydroxytyrosol) Mitigates the Increase in Spontaneous Oxidation of Dopamine During Monoamine Oxidase Inhibition in PC12 Cells. Neurochem. Res. 2016, 41, 2173–2178.
- Pérez-Barrón, G.; Montes, S.; Rubio-Osornio, M.; Avila-Acevedo, J.G.; Garcia-Jimenez, S.; Rios, L.C.; Monroy-Noyola, A. Hydroxytyrosol inhibits MAO isoforms and prevents neurotoxicity inducible by MPP+ in vivo. Front. Biosci. 2020, 12, 25–37.
- Pérez-Barrón, G.; Montes, S.; Aguirre-Vidal, Y.; Santiago, M.; Gallardo, E.; Espartero, J.L.; Ríos, C.; Monroy-Noyola, A. Antioxidant Effect of Hydroxytyrosol, Hydroxytyrosol Acetate and Nitrohydroxytyrosol in a Rat MPP+ Model of Parkinson’s Disease. Neurochem. Res. 2021, 46, 2923–2935.
- Singh, R.; Zahra, W.; Singh, S.S.; Birla, H.; Rathore, A.S.; Keshri, P.K.; Dilnashin, H.; Singh, S.; Singh, S.P. Oleuropein confers neuroprotection against rotenone-induced model of Parkinson’s disease via BDNF/CREB/Akt pathway. Sci. Rep. 2023, 13, 2452.
- Palazzi, L.; Leri, M.; Cesaro, S.; Stefani, M.; Bucciantini, M.; de Laureto, P.P. Insight into the molecular mechanism underlying the inhibition of α-synuclein aggregation by hydroxytyrosol. Biochem. Pharmacol. 2020, 173, 113722.
- Mohammad-Beigi, H.; Aliakbari, F.; Sahin, C.; Lomax, C.; Tawfike, A.; Schafer, N.P.; Amiri-Nowdijeh, A.; Eskandari, H.; Møller, I.M.; Hosseini-Mazinani, M.; et al. Oleuropein derivatives from olive fruit extracts reduce α-synuclein fibrillation and oligomer toxicity. J. Biol. Chem. 2019, 294, 4215–4232.
- Borah, P.; Sanjeev, A.; Mattaparthi, V.S.K. Computational investigation on the effect of Oleuropein aglycone on the α-synuclein aggregation. J. Biomol. Struct. Dyn. 2020, 39, 1259–1270.
- Brunetti, G.; Di Rosa, G.; Scuto, M.; Leri, M.; Stefani, M.; Schmitz-Linneweber, C.; Calabrese, V.; Saul, N. Healthspan Maintenance and Prevention of Parkinson’s-like Phenotypes with Hydroxytyrosol and Oleuropein Aglycone in C. elegans. Int. J. Mol. Sci. 2020, 21, 2588.
- Di Rosa, G.; Brunetti, G.; Scuto, M.; Trovato Salinaro, A.; Calabrese, E.J.; Crea, R.; Schmitz-Linneweber, C.; Calabrese, V.; Saul, N. Health-span Enhancement by Olive Polyphenols in C. elegans Wild Type and Parkinson’s Models. Int. J. Mol. Sci. 2020, 21, 3893.
- Marks, J.L.; Porte, D.; Stahl, W.L.; Baskin, D.G. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology 1990, 127, 3234–3236.
- Okouchi, M.; Okayama, N.; Alexander, J.S.; Aw, T.Y. NRF2-Dependent Glutamate-L-Cysteine Ligase Catalytic Subunit Expression Mediates Insulin Protection Against Hyperglycemia-Induced Brain Endothelial Cell Apoptosis. Curr. Neurovascular Res. 2006, 3, 249–261.
- Hoyer, S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 2004, 490, 115–125.
- De La Monte, S.M. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimers Disease. Curr. Alzheimer Res. 2012, 9, 35–66.
- Zheng, A.; Li, H.; Xu, J.; Cao, K.; Li, H.; Pu, W.; Yang, Z.; Peng, Y.; Long, J.; Liu, J.; et al. Hydroxytyrosol improves mitochondrial function and reduces oxidative stress in the brain of db/db mice: Role of AMP-activated protein kinase activation. Br. J. Nutr. 2015, 113, 1667–1676.
- Cantó, C.; Auwerx, J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Infect. Dis. 2009, 20, 98–105.
- Chaari, A. Inhibition of human islet amyloid polypeptide aggregation and cellular toxicity by oleuropein and derivatives from olive oil. Int. J. Biol. Macromol. 2020, 162, 284–300.
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215.
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380.
- Wu, J.J.; Liu, J.; Chen, E.B.; Wang, J.J.; Cao, L.; Narayan, N.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. Increased Mammalian Lifespan and a Segmental and Tissue-Specific Slowing of Aging after Genetic Reduction of mTOR Expression. Cell Rep. 2013, 4, 913–920.
- Zarzuelo, M.J.; López-Sepúlveda, R.; Sánchez, M.; Romero, M.; Gómez-Guzmán, M.; Ungvary, Z.; Pérez-Vizcaíno, F.; Jiménez, R.; Duarte, J. SIRT1 inhibits NADPH oxidase activation and protects endothelial function in the rat aorta: Implications for vascular aging. Biochem. Pharmacol. 2013, 85, 1288–1296.
- Shang, J.; Che, S.; Zhu, M. Oleuropein Improves Cognitive Dysfunction and Neuroinflammation in Diabetic Rats through the PI3K/Akt/mTOR Pathway. Appl. Bionics Biomech. 2022, 5892463.
- Zheng, S.; Huang, K.; Tong, T. Efficacy and Mechanisms of Oleuropein in Mitigating Diabetes and Diabetes Complications. J. Agric. Food Chem. 2021, 69, 6145–6155.
- Benlarbi, M.; Jemai, H.; Hajri, K.; Mbarek, S.; Amri, E.; Jebbari, M.; Hammoun, I.; Baccouche, B.; Mihoubi, N.B.; Zemmal, A.; et al. Neuroprotective effects of oleuropein on retina photoreceptors cells primary culture and olive leaf extract and oleuropein inhibitory effects on aldose reductase in a diabetic model: Meriones shawi. Arch. Physiol. Biochem. 2020, 128, 593–600.
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40.
- Karussis, D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: A critical review. J. Autoimmun. 2014, 48–49, 134–142.
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal Transection in the Lesions of Multiple Sclerosis. N. Engl. J. Med. 1998, 338, 278–285.
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705.
- Conde, C.; Escribano, B.; Luque, E.; Aguilar-Luque, M.; Feijóo, M.; Ochoa, J.; Latorre, M.; Giraldo, A.; Lillo, R.; Aguera-Morales, E.; et al. The protective effect of extra-virgin olive oil in the experimental model of multiple sclerosis in the rat. Nutr. Neurosci. 2018, 23, 37–48.
- Liuzzi, G.M.; Latronico, T.; Branà, M.T.; Gramegna, P.; Coniglio, M.G.; Rossano, R.; Larocca, M.; Riccio, P. Structure-Dependent Inhibition of Gelatinases by Dietary Antioxidants in Rat Astrocytes and Sera of Multiple Sclerosis Patients. Neurochem. Res. 2011, 36, 518–527.
- Giacometti, J.; Grubić-Kezele, T. Olive Leaf Polyphenols Attenuate the Clinical Course of Experimental Autoimmune Encephalomyelitis and Provide Neuroprotection by Reducing Oxidative Stress, Regulating Microglia and SIRT1, and Preserving Myelin Integrity. Oxidative Med. Cell. Longev. 2020, 2020, 1–20.
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228.
- Cameron, H.A.; Woolley, C.S.; McEwen, B.S.; Gould, E. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 1993, 56, 337–344.
- Kempermann, G.; Song, H.; Gage, F.H. Neurogenesis in the Adult Hippocampus. Cold Spring Harb. Perspect. Biol. 2015, 7, a018812.
- Lim, D.A.; Alvarez-Buylla, A. The Adult Ventricular–Subventricular Zone (V-SVZ) and Olfactory Bulb (OB) Neurogenesis. Cold Spring Harb. Perspect. Biol. 2016, 8, a018820.
- Aimone, J.B.; Gage, F.H. Modeling new neuron function: A history of using computational neuroscience to study adult neurogenesis. Eur. J. Neurosci. 2011, 33, 1160–1169.
- Farioli-Vecchioli, S.; Saraulli, D.; Costanzi, M.; Pacioni, S.; Cinà, I.; Aceti, M.; Micheli, L.; Bacci, A.; Cestari, V.; Tirone, F. The Timing of Differentiation of Adult Hippocampal Neurons Is Crucial for Spatial Memory. PLoS Biol. 2008, 6, e246.
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 2018, 22, 589–599.e5.
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381.
- Bonaguidi, M.A.; Wheeler, M.A.; Shapiro, J.S.; Stadel, R.P.; Sun, G.J.; Ming, G.L.; Song, H. In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 2011, 145, 1142–1155.
- Urbán, N.; Berg, D.L.C.V.D.; Forget, A.; Andersen, J.; Demmers, J.A.A.; Hunt, C.; Ayrault, O.; Guillemot, F. Return to quiescence of mouse neural stem cells by degradation of a proactivation protein. Science 2016, 353, 292–295.
- Obernier, K.; Cebrian-Silla, A.; Thomson, M.; Parraguez, J.I.; Anderson, R.; Guinto, C.; Rodriguez, J.R.; Garcia-Verdugo, J.-M.; Alvarez-Buylla, A. Adult Neurogenesis Is Sustained by Symmetric Self-Renewal and Differentiation. Cell Stem Cell 2018, 22, 221–234.e8.
- Pilz, G.-A.; Bottes, S.; Betizeau, M.; Jörg, D.J.; Carta, S.; Simons, B.D.; Helmchen, F.; Jessberger, S. Live imaging of neurogenesis in the adult mouse hippocampus. Science 2018, 359, 658–662.
- Encinas, J.M.; Michurina, T.V.; Peunova, N.; Park, J.-H.; Tordo, J.; Peterson, D.A.; Fishell, G.; Koulakov, A.; Enikolopov, G. Division-Coupled Astrocytic Differentiation and Age-Related Depletion of Neural Stem Cells in the Adult Hippocampus. Cell Stem Cell 2011, 8, 566–579.
- Kuhn, H.-G.; Dickinson-Anson, H.; Gage, F.H. Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. J. Neurosci. 1996, 16, 2027–2033.
- van Praag, H.; Shubert, T.; Zhao, C.; Gage, F.H. Exercise Enhances Learning and Hippocampal Neurogenesis in Aged Mice. J. Neurosci. 2005, 25, 8680–8685.
- van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266–270.
- Siette, J.; Westbrook, R.F.; Cotman, C.; Sidhu, K.; Zhu, W.; Sachdev, P.; Valenzuela, M.J. Age-Specific Effects of Voluntary Exercise on Memory and the Older Brain. Biol. Psychiatry 2013, 73, 435–442.
- Micheli, L.; D’Andrea, G.; Ceccarelli, M.; Ferri, A.; Scardigli, R.; Tirone, F. p16Ink4a Prevents the Activation of Aged Quiescent Dentate Gyrus Stem Cells by Physical Exercise. Front. Cell. Neurosci. 2019, 13, 10.
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of Hippocampal Neurogenesis for the Behavioral Effects of Antidepressants. Science 2003, 301, 805–809.
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic Antidepressant Treatment Increases Neurogenesis in Adult Rat Hippocampus. J. Neurosci. 2000, 20, 9104–9110.
- Couillard-Despres, S.; Wuertinger, C.; Kandasamy, M.; Caioni, M.; Stadler, K.; Aigner, R.; Bogdahn, U.; Aigner, L. Ageing abolishes the effects of fluoxetine on neurogenesis. Mol. Psychiatry 2009, 14, 856–864.
- Gould, E.; Beylin, A.V.; Tanapat, P.; Reeves, A.J.; Shors, T.J. Learning enhances adult neurogenesis in the hippocampal formation. Nat. Neurosci. 1999, 2, 260–265.
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. Experience-Induced Neurogenesis in the Senescent Dentate Gyrus. J. Neurosci. 1998, 18, 3206–3212.
- Encinas, J.M.; Vaahtokari, A.; Enikolopov, G. Fluoxetine targets early progenitor cells in the adult brain. Proc. Natl. Acad. Sci. USA 2006, 103, 8233–8238.
- Micheli, L.; Ceccarelli, M.; D’Andrea, G.; Costanzi, M.; Giacovazzo, G.; Coccurello, R.; Caruso, C.; Tirone, F. Fluoxetine or Sox2 reactivate proliferation-defective stem and progenitor cells of the adult and aged dentate gyrus. Neuropharmacology 2018, 141, 316–330.
- Micheli, L.; Ceccarelli, M.; D’Andrea, G.; Tirone, F. Depression and adult neurogenesis: Positive effects of the antidepressant fluoxetine and of physical exercise. Brain Res. Bull. 2018, 143, 181–193.
- Zhou, W.B.; Miao, Z.N.; Zhang, B.; Long, W.; Zheng, F.X.; Kong, J.; Yu, B. Luteolin induces hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neural Regen Res. 2019, 4, 613–620.
- Ceccarelli, M.; D’Andrea, G.; Micheli, L.; Tirone, F. Interaction Between Neurogenic Stimuli and the Gene Network Controlling the Activation of Stem Cells of the Adult Neurogenic Niches, in Physiological and Pathological Conditions. Front. Cell Dev. Biol. 2020, 8, 211.
- Zheng, A.; Li, H.; Cao, K.; Xu, J.; Zou, X.; Li, Y.; Chen, C.; Liu, J.; Feng, Z. Maternal hydroxytyrosol administration improves neurogenesis and cognitive function in prenatally stressed offspring. J. Nutr. Biochem. 2015, 26, 190–199.
- Calahorra, J.; Shenk, J.; Wielenga, V.H.; Verweij, V.; Geenen, B.; Dederen, P.J.; Peinado, M.; Siles, E.; Wiesmann, M.; Kiliaan, A.J. Hydroxytyrosol, the Major Phenolic Compound of Olive Oil, as an Acute Therapeutic Strategy after Ischemic Stroke. Nutrients 2019, 11, 2430.
- Mnafgui, K.; Ghazouani, L.; Hajji, R.; Tlili, A.; Derbali, F.; da Silva, F.I.; Araújo, J.L.; Schinoff, B.D.O.; Bachega, J.F.R.; Santos, A.L.D.S.; et al. Oleuropein Protects Against Cerebral Ischemia Injury in Rats: Molecular Docking, Biochemical and Histological Findings. Neurochem. Res. 2021, 2131–2142.
- Yu, H.; Liu, P.; Tang, H.; Jing, J.; Lv, X.; Chen, L.; Jiang, L.; Xu, J.; Li, J. Oleuropein, a natural extract from plants, offers neuroprotection in focal cerebral ischemia/reperfusion injury in mice. Eur. J. Pharmacol. 2016, 775, 113–119.
- Shi, J.; Wu, G.; Zou, X.; Jiang, K. Oleuropein protects intracerebral hemorrhage-induced disruption of blood-brain barrier through alleviation of oxidative stress. Pharmacol. Rep. 2017, 69, 1206–1212.
- Warner-Schmidt, J.L.; Duman, R.S. Hippocampal neurogenesis: Opposing effects of stress and antidepressant treatment. Hippocampus 2006, 16, 239–249.
- Duman, C.H.; Schlesinger, L.; Russell, D.S.; Duman, R.S. Voluntary exercise produces antidepressant and anxiolytic behavioral effects in mice. Brain Res. 2008, 1199, 148–158.
- Kheirbek, M.A.; Drew, L.J.; Burghardt, N.S.; Costantini, D.O.; Tannenholz, L.; Ahmari, S.E.; Zeng, H.; Fenton, A.A.; Hen, R. Differential Control of Learning and Anxiety along the Dorsoventral Axis of the Dentate Gyrus. Neuron 2013, 77, 955–968.
- David, D.J.; Samuels, B.A.; Rainer, Q.; Wang, J.-W.; Marsteller, D.; Mendez, I.; Drew, M.; Craig, D.A.; Guiard, B.P.; Guilloux, J.-P.; et al. Neurogenesis-Dependent and -Independent Effects of Fluoxetine in an Animal Model of Anxiety/Depression. Neuron 2009, 62, 479–493.
- Alexopoulos, G.S. Mechanisms and treatment of late-life depression. Transl. Psychiatry 2019, 9, 188.
- McAvoy, K.; Russo, C.; Kim, S.; Rankin, G.; Sahay, A. Fluoxetine induces input-specific hippocampal dendritic spine remodeling along the septotemporal axis in adulthood and middle age. Hippocampus 2015, 25, 1429–1446.
- Zhao, Y.-T.; Zhang, L.; Yin, H.; Shen, L.; Zheng, W.; Zhang, K.; Zeng, J.; Hu, C.; Liu, Y. Hydroxytyrosol alleviates oxidative stress and neuroinflammation and enhances hippocampal neurotrophic signaling to improve stress-induced depressive behaviors in mice. Food Funct. 2021, 12, 5478–5487.
- Marlatt, M.W.; Potter, M.C.; Lucassen, P.J.; van Praag, H. Running throughout middle-age improves memory function, hippocampal neurogenesis, and BDNF levels in female C57BL/6J mice. Dev. Neurobiol. 2012, 72, 943–952.
- Fan, L.; Peng, Y.; Wang, J.; Ma, P.; Zhao, L.; Li, X. Total glycosides from stems of Cistanche tubulosa alleviate depression-like behaviors: Bidirectional interaction of the phytochemicals and gut microbiota. Phytomedicine 2021, 83, 153471.
- Belzung, C.; de Villemeur, E.B. The design of new antidepressants: Can formal models help? A first attempt using a model of the hippocampal control over the HPA-axis based on a review from the literature. Behav. Pharmacol. 2010, 21, 677–689.
- Alenina, N.; Klempin, F. The role of serotonin in adult hippocampal neurogenesis. Behav. Brain Res. 2015, 277, 49–57.
- Lee, B.; Shim, I.; Lee, H.; Hahm, D.-H. Oleuropein reduces anxiety-like responses by activating of serotonergic and neuropeptide Y (NPY)-ergic systems in a rat model of post-traumatic stress disorder. Anim. Cells Syst. 2018, 22, 109–117.
- Badr, A.M.; Attia, H.A.; Al-Rasheed, N. Oleuropein Reverses Repeated Corticosterone-Induced Depressive-Like Behavior in mice: Evidence of Modulating Effect on Biogenic Amines. Sci. Rep. 2020, 10, 3336.
- Tinnirello, A.; Mazzoleni, S.; Santi, C. Chronic Pain in the Elderly: Mechanisms and Distinctive Features. Biomolecules 2021, 11, 1256.
- Yu, H.; Zhang, Z.; Wei, F.; Hou, G.; You, Y.; Wang, X.; Cao, S.; Yang, X.; Liu, W.; Zhang, S.; et al. Hydroxytyrosol Ameliorates Intervertebral Disc Degeneration and Neuropathic Pain by Reducing Oxidative Stress and Inflammation. Oxidative Med. Cell. Longev. 2022, 2022, 2240894.
- Kamil, K.; Yazid, M.D.; Idrus, R.B.H.; Kumar, J. Hydroxytyrosol Promotes Proliferation of Human Schwann Cells: An In Vitro Study. Int. J. Environ. Res. Public Health 2020, 17, 4404.
- Ristagno, G.; Fumagalli, F.; Porretta-Serapiglia, C.; Orrù, A.; Cassina, C.; Pesaresi, M.; Masson, S.; Villanova, L.; Merendino, A.; Villanova, A.; et al. Hydroxytyrosol Attenuates Peripheral Neuropathy in Streptozotocin-Induced Diabetes in Rats. J. Agric. Food Chem. 2012, 60, 5859–5865.
- Khalatbary, A.R.; Ahmadvand, H. Effect of Oleuropein on Tissue Myeloperoxidase Activity in Experimental Spinal Cord Trauma. Iran. Biomed. J. 2011, 15, 164–167.
- Zhang, Y.-J.; Chen, X.; Zhang, L.; Li, J.; Li, S.-B.; Zhang, X.; Qin, L.; Sun, F.-R.; Li, D.-Q.; Ding, G.-Z. Protective effects of 3,4-dihydroxyphenylethanol on spinal cord injury-induced oxidative stress and inflammation. Neuroreport 2019, 30, 1016–1024.
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2020, 22, 75–95.
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453.
- Ungerleider, K.; Beck, J.; Lissa, D.; Turnquist, C.; Horikawa, I.; Harris, B.T.; Harris, C.C. Astrocyte senescence and SASP in neurodegeneration: Tau joins the loop. Cell Cycle 2021, 20, 752–764.
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453.
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189.
- Menicacci, B.; Cipriani, C.; Margheri, F.; Mocali, A.; Giovannelli, L. Modulation of the Senescence-Associated Inflammatory Phenotype in Human Fibroblasts by Olive Phenols. Int. J. Mol. Sci. 2017, 18, 2275.
- Frediani, E.; Scavone, F.; Laurenzana, A.; Chillà, A.; Tortora, K.; Cimmino, I.; Leri, M.; Bucciantini, M.; Mangoni, M.; Fibbi, G.; et al. Olive phenols preserve lamin B1 expression reducing cGAS/STING/NFκB-mediated SASP in ionizing radiation-induced senescence. J. Cell. Mol. Med. 2022, 26, 2337–2350.
- Jeon, S.; Choi, M. Anti-inflammatory and anti-aging effects of hydroxytyrosol on human dermal fibroblasts (HDFs). Biomed. Dermatol. 2018, 2, 21.
- Varela-Eirín, M.; Carpintero-Fernández, P.; Sánchez-Temprano, A.; Varela-Vázquez, A.; Paíno, C.L.; Casado-Díaz, A.; Continente, A.C.; Mato, V.; Fonseca, E.; Kandouz, M.; et al. Senolytic activity of small molecular polyphenols from olive restores chondrocyte redifferentiation and promotes a pro-regenerative environment in osteoarthritis. Aging 2020, 12, 15882–15905.
- Dolivo, D.; Hernandez, S.; Dominko, T. Cellular lifespan and senescence: A complex balance between multiple cellular pathways. Bioessays 2016, 38, S33–S44.
- Katsiki, M.; Chondrogianni, N.; Chinou, I.; Rivett, A.J.; Gonos, E.S. The Olive Constituent Oleuropein Exhibits Proteasome Stimulatory Properties In Vitro and Confers Life Span Extension of Human Embryonic Fibroblasts. Rejuvenation Res. 2007, 10, 157–172.
- Sarsour, E.H.; Kumar, M.G.; Kalen, A.L.; Goswami, M.; Buettner, G.R.; Goswami, P.C. MnSOD activity regulates hydroxytyrosol-induced extension of chronological lifespan. Age 2011, 34, 95–109.
- Feng, S.; Zhang, C.; Chen, T.; Zhou, L.; Huang, Y.; Yuan, M.; Li, T.; Ding, C. Oleuropein Enhances Stress Resistance and Extends Lifespan via Insulin/IGF-1 and SKN-1/Nrf2 Signaling Pathway in Caenorhabditis elegans. Antioxidants 2021, 10, 1697.
- Romero-Márquez, J.M.; Navarro-Hortal, M.D.; Jiménez-Trigo, V.; Muñoz-Ollero, P.; Forbes-Hernández, T.Y.; Esteban-Muñoz, A.; Giampieri, F.; Noya, I.D.; Bullón, P.; Vera-Ramírez, L.; et al. An Olive-Derived Extract 20% Rich in Hydroxytyrosol Prevents β-Amyloid Aggregation and Oxidative Stress, Two Features of Alzheimer Disease, via SKN-1/NRF2 and HSP-16.2 in Caenorhabditis elegans. Antioxidants 2022, 11, 629.
- Pedersen, B.K. Physical activity and muscle–brain crosstalk. Nat. Rev. Endocrinol. 2019, 15, 383–392.
- Scisciola, L.; Fontanella, R.; Surina; Cataldo, V.; Paolisso, G.; Barbieri, M. Sarcopenia and Cognitive Function: Role of Myokines in Muscle Brain Cross-Talk. Life 2021, 11, 173.
- Greco, E.A.; Pietschmann, P.; Migliaccio, S. Osteoporosis and Sarcopenia Increase Frailty Syndrome in the Elderly. Front. Endocrinol. 2019, 10, 255.
- Demontis, F.; Piccirillo, R.; Goldberg, A.L.; Perrimon, N. The influence of skeletal muscle on systemic aging and lifespan. Aging Cell 2013, 12, 943–949.
- Szentesi, P.; Csernoch, L.; Dux, L.; Keller-Pintér, A. Changes in Redox Signaling in the Skeletal Muscle with Aging. Oxid. Med. Cell. Longev. 2019, 4617801.
- Foreman, N.A.; Hesse, A.S.; Ji, L.L. Redox Signaling and Sarcopenia: Searching for the Primary Suspect. Int. J. Mol. Sci. 2021, 22, 9045.
- Pierno, S.; Tricarico, D.; Liantonio, A.; Mele, A.; Digennaro, C.; Rolland, J.-F.; Bianco, G.; Villanova, L.; Merendino, A.; Camerino, G.M.; et al. An olive oil-derived antioxidant mixture ameliorates the age-related decline of skeletal muscle function. AGE 2014, 36, 73–88.
- El, S.N.; Karakaya, S. Olive tree (Olea europaea) leaves: Potential beneficial effects on human health. Nutr. Rev. 2009, 67, 632–638.
- González-Hedström, D.; Priego, T.; Amor, S.; de la Fuente-Fernández, M.; Martín, A.; López-Calderón, A.; Inarejos-García, A.; García-Villalón, Á.L.; Granado, M. Olive Leaf Extract Supplementation to Old Wistar Rats Attenuates Aging-Induced Sarcopenia and Increases Insulin Sensitivity in Adipose Tissue and Skeletal Muscle. Antioxidants 2021, 10, 737.
- Gumucio, J.P.; Mendias, C.L. Atrogin-1, MuRF-1, and sarcopenia. Endocrine 2012, 43, 12–21.
- Ryan, A.S.; Li, G. Skeletal muscle myostatin gene expression and sarcopenia in overweight and obese middle-aged and older adults. JCSM Clin. Rep. 2021, 6, 137–142.
- Lee, S.-J. Targeting the myostatin signaling pathway to treat muscle loss and metabolic dysfunction. J. Clin. Investig. 2021, 131, e148372.
- Luo, L.; Martin, S.C.; Parkington, J.; Cadena, S.M.; Zhu, J.; Ibebunjo, C.; Summermatter, S.; Londraville, N.; Patora-Komisarska, K.; Widler, L.; et al. HDAC4 Controls Muscle Homeostasis through Deacetylation of Myosin Heavy Chain, PGC-1α, and Hsc70. Cell Rep. 2019, 29, 749–763.e12.
- Dedkov, E.I.; Kostrominova, T.Y.; Borisov, A.B.; Carlson, B.M. MyoD and myogenin protein expression in skeletal muscles of senile rats. Cell Tissue Res. 2003, 311, 401–416.
- González-Hedström, D.; de la Fuente-Fernández, M.; Priego, T.; Martín, A.; Amor, S.; López-Calderón, A.; Inarejos-García, A.; García-Villalón, Á.L.; Granado, M. Addition of Olive Leaf Extract to a Mixture of Algae and Extra Virgin Olive Oils Decreases Fatty Acid Oxidation and Synergically Attenuates Age-Induced Hypertension, Sarcopenia and Insulin Resistance in Rats. Antioxidants 2021, 10, 1066.
- González-Hedström, D.; Priego, T.; López-Calderón, A.; Amor, S.; de la Fuente-Fernández, M.; Inarejos-García, A.M.; Gar-cía-Villalón, L.; Martín, A.I.; Granado, M. Beneficial Effects of a Mixture of Algae and Extra Virgin Olive Oils on the Age-Induced Alterations of Rodent Skeletal Muscle: Role of HDAC-4. Nutrients 2020, 13, 44.
- González-Hedström, D.; Amor, S.; de la Fuente-Fernández, M.; Tejera-Muñoz, A.; Priego, T.; Martín, A.I.; López-Calderón, A.; Inarejos-García, A.M.; García-Villalón, L.; Granado, M. A Mixture of Algae and Extra Virgin Olive Oils Attenuates the Cardiometabolic Alterations Associated with Aging in Male Wistar Rats. Antioxidants 2020, 9, 483.
- Reginster, J.-Y.; Beaudart, C.; Buckinx, F.; Bruyère, O. Osteoporosis and Sarcopenia: Two Diseases or One? Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 31–36.
- Clynes, M.A.; Gregson, C.L.; Bruyère, O.; Cooper, C.; Dennison, E.M. Osteosarcopenia: Where osteoporosis and sarcopenia collide. Rheumatology 2021, 60, 529–537.
- Chin, K.-Y.; Ima-Nirwana, S. Olives and Bone: A Green Osteoporosis Prevention Option. Int. J. Environ. Res. Public Health 2016, 13, 755.
- Nicolin, V.; de Tommasi, N.; Nori, S.L.; Costantinides, F.; Berton, F.; di Lenarda, R. Modulatory Effects of Plant Polyphenols on Bone Remodeling: A Prospective View From the Bench to Bedside. Front. Endocrinol. 2019, 10, 494.
- Hagiwara, K.; Goto, T.; Araki, M.; Miyazaki, H.; Hagiwara, H. Olive polyphenol hydroxytyrosol prevents bone loss. Eur. J. Pharmacol. 2011, 662, 78–84.
- Puel, C.; Mathey, J.; Agalias, A.; Kati-Coulibaly, S.; Mardon, J.; Obled, C.; Davicco, M.-J.; Lebecque, P.; Horcajada, M.-N.; Skaltsounis, A.L.; et al. Dose–response study of effect of oleuropein, an olive oil polyphenol, in an ovariectomy/inflammation experimental model of bone loss in the rat. Clin. Nutr. 2006, 25, 859–868.
- Puel, C.; Mardon, J.; Agalias, A.; Davicco, M.-J.; Lebecque, P.; Mazur, A.; Horcajada, M.-N.; Skaltsounis, A.-L.; Coxam, V. Major Phenolic Compounds in Olive Oil Modulate Bone Loss in an Ovariectomy/Inflammation Experimental Model. J. Agric. Food Chem. 2008, 56, 9417–9422.
- Liu, H.; Zhao, A.; Huang, Y.; Hou, A.; Miao, W.; Hong, L.; Deng, N.; Fan, Y. Efficacy and Mechanisms of Oleurpein in Postmenopausal Osteoporosis. Comput. Math. Methods Med. 2022, 2022, 9767113.
- Cai, W.J.; Chen, Y.; Shi, L.X.; Cheng, H.R.; Banda, I.; Ji, Y.H.; Wang, Y.T.; Li, X.M.; Mao, Y.X.; Zhang, D.F.; et al. AKT-GSK3βSignaling Pathway Regulates Mitochondrial Dysfunction-Associated OPA1 Cleavage Contributing to Osteoblast Apoptosis: Preventative Effects of Hydroxytyrosol. Oxidative Med. Cell. Longev. 2019, 2019, 4101738.
- Powers, S.K.; Schrager, M. Redox signaling regulates skeletal muscle remodeling in response to exercise and prolonged inactivity. Redox Biol. 2022, 54, 102374.
- Liu, J.; Peng, Y.; Feng, Z.; Shi, W.; Qu, L.; Li, Y.; Liu, J.; Long, J. Reloading functionally ameliorates disuse-induced muscle atrophy by reversing mitochondrial dysfunction, and similar benefits are gained by administering a combination of mitochondrial nutrients. Free. Radic. Biol. Med. 2014, 69, 116–128.
- Wang, F.; Wang, X.; Liu, Y.; Zhang, Z. Effects of Exercise-Induced ROS on the Pathophysiological Functions of Skeletal Muscle. Oxidative Med. Cell. Longev. 2021, 2021, 3846122.
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425.
- Feng, Z.; Bai, L.; Yan, J.; Li, Y.; Shen, W.; Wang, Y.; Wertz, K.; Weber, P.; Zhang, Y.; Chen, Y.; et al. Mitochondrial dynamic remodeling in strenuous exercise-induced muscle and mitochondrial dysfunction: Regulatory effects of hydroxytyrosol. Free. Radic. Biol. Med. 2011, 50, 1437–1446.
- Wang, X.; Li, H.; Zheng, A.; Yang, L.; Liu, J.; Chen, C.; Tang, Y.; Zou, X.; Li, Y.; Long, J.; et al. Mitochondrial dysfunction-associated OPA1 cleavage contributes to muscle degeneration: Preventative effect of hydroxytyrosol acetate. Cell Death Dis. 2014, 5, e1521.
- Friedel, A.; Raederstor, D.; Roos, F.; Toepfer, C.; Wertz, K. Hydroxytyrosol Benefits Muscle Differentiation and Muscle Contrac-tion and Relaxation. U.S. Patent 13/550,972, 7 March 2013.
- Kikusato, M.; Muroi, H.; Uwabe, Y.; Furukawa, K.; Toyomizu, M. Oleuropein induces mitochondrial biogenesis and decreases reactive oxygen species generation in cultured avian muscle cells, possibly via an up-regulation of peroxisome prolferator-activated receptor γ coactivator-1α. Anim. Sci. J. 2016, 87, 1371–1378.
- Muroi, H.; Hori, K.; Tokutake, Y.; Hakamata, Y.; Kawabata, F.; Toyomizu, M.; Kikusato, M. Oleuropein suppresses mitochondrial reactive oxygen species generation possibly via an activation of transient receptor potential V1 and sirtuin-1 in cultured chicken muscle cells. Anim. Sci. J. 2022, 93, e13677.
- Marcelo, K.L.; Means, A.R.; York, B. The Ca 2+ /Calmodulin/CaMKK2 Axis: Nature’s Metabolic CaMshaft. Trends Endocrinol. Metab. 2016, 27, 706–718.
- Shou, J.; Chen, P.-J.; Xiao, W.-H. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol. Metab. Syndr. 2020, 12, 14.
- Nishikawa, H.; Asai, A.; Fukunishi, S.; Nishiguchi, S.; Higuchi, K. Metabolic Syndrome and Sarcopenia. Nutrients 2021, 13, 3519.
- Yeo, Y.H.; Lai, Y.-C. Redox regulation of metabolic syndrome: Recent developments in skeletal muscle insulin resistance and non-alcoholic fatty liver disease (NAFLD). Curr. Opin. Physiol. 2019, 9, 79–86.
- Teng, S.; Huang, P. The effect of type 2 diabetes mellitus and obesity on muscle progenitor cell function. Stem Cell Res. Ther. 2019, 10, 103.
- Hirata, Y.; Nomura, K.; Senga, Y.; Okada, Y.; Kobayashi, K.; Okamoto, S.; Minokoshi, Y.; Imamura, M.; Takeda, S.; Hosooka, T.; et al. Hyperglycemia induces skeletal muscle atrophy via a WWP1/KLF15 axis. J. Clin. Investig. 2019, 4, e124952.
- Fellmann, L.; Nascimento, A.R.; Tibiriça, E.; Bousquet, P. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol. Ther. 2013, 137, 331–340.
- Cao, K.; Xu, J.; Zou, X.; Li, Y.; Chen, C.; Zheng, A.; Li, H.; Li, H.; Szeto, I.M.-Y.; Shi, Y.; et al. Hydroxytyrosol prevents diet-induced metabolic syndrome and attenuates mitochondrial abnormalities in obese mice. Free. Radic. Biol. Med. 2014, 67, 396–407.
- Drira, R.; Sakamoto, K. Modulation of adipogenesis, lipolysis and glucose consumption in 3T3-L1 adipocytes and C2C12 myotubes by hydroxytyrosol acetate: A comparative study. Biochem. Biophys. Res. Commun. 2013, 440, 576–581.
- Fujiwara, Y.; Tsukahara, C.; Ikeda, N.; Sone, Y.; Ishikawa, T.; Ichi, I.; Koike, T.; Aoki, Y. Oleuropein improves insulin resistance in skeletal muscle by promoting the translocation of GLUT4. J. Clin. Biochem. Nutr. 2017, 61, 196–202.
- Hadrich, F.; Garcia, M.; Maalej, A.; Moldes, M.; Isoda, H.; Feve, B.; Sayadi, S. Oleuropein activated AMPK and induced insulin sensitivity in C2C12 muscle cells. Life Sci. 2016, 151, 167–173.
- Alkhateeb, H.H.; Kaplan, N.M.; Al-Duais, M. Understanding the Mechanism Underlie the Antidiabetic Activity of Oleuropein Using Ex-Vivo Approach. Rep. Biochem. Mol. Biol. 2022, 11, 146–156.
- Giacometti, J.; Muhvić, D.; Grubić-Kezele, T.; Nikolić, M.; Šoić-Vranić, T.; Bajek, S.; Muhvić, D. Olive Leaf Polyphenols (OLPs) Stimulate GLUT4 Expression and Translocation in the Skeletal Muscle of Diabetic Rats. Int. J. Mol. Sci. 2020, 21, 8981.
- Jaldin-Fincati, J.R.; Pavarotti, M.; Frendo-Cumbo, S.; Bilan, P.J.; Klip, A. Update on GLUT4 Vesicle Traffic: A Cornerstone of Insulin Action. Trends Endocrinol. Metab. 2017, 28, 597–611.
- Van de Guchte, M.; Blottiere, H.; Doré, J. Humans as holobionts: Implications for prevention and therapy. Microbiome 2018, 6, 81.
- Milani, C.; Duranti, S.; Bottacini, F.; Casey, E.; Turroni, F.; Mahony, J.; Belzer, C.; Delgado Palacio, S.; Arboleya Montes, S.; Mancabelli, L.; et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev. 2017, 81, e00036-17.
- Sanchez-Morate, E.; Gimeno-Mallench, L.; Stromsnes, K.; Sanz-Ros, J.; Román-Domínguez, A.; Parejo-Pedrajas, S.; Inglés, M.; Olaso, G.; Gambini, J.; Mas-Bargues, C. Relationship between Diet, Microbiota, and Healthy Aging. Biomedicines 2020, 8, 287.
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51.
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. 2019, 76, 473–493.
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14.
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An Obesity-Associated Gut Microbiome with Increased Capacity for Energy Harvest. Nature 2006, 444, 1027–1031.
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14.
- Thaiss, C.A.; Itav, S.; Rothschild, D.; Meijer, M.T.; Levy, M.; Moresi, C.; Dohnalová, L.; Braverman, S.; Rozin, S.; Malitsky, S.; et al. Persistent microbiome alterations modulate the rate of post-dieting weight regain. Nature 2016, 540, 544–551.
- Chen, Y.; Zhou, J.; Wang, L. Role and Mechanism of Gut Microbiota in Human Disease. Front. Cell. Infect. Microbiol. 2021, 11, 625913.
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimaraes, V.D.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.-P. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009, 9, 123.
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013.
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie van Leeuwenhoek 2020, 113, 2019–2040.
- Derrien, M.; Alvarez, A.-S.; de Vos, W.M. The Gut Microbiota in the First Decade of Life. Trends Microbiol. 2019, 27, 997–1010.
- Del Chierico, F.; Vernocchi, P.; Petrucca, A.; Paci, P.; Fuentes, S.; Praticò, G.; Capuani, G.; Masotti, A.; Reddel, S.; Russo, A.; et al. Phylogenetic and Metabolic Tracking of Gut Microbiota during Perinatal Development. PLoS ONE 2015, 10, e0137347.
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227.
- Claesson, M.J.; Cusack, S.; O’Sullivan, O.; Greene-Diniz, R.; De Weerd, H.; Flannery, E.; Marchesi, J.R.; Falush, D.; Dinan, T.G.; Fitzgerald, G.F.; et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc. Natl. Acad. Sci. USA 2011, 108, 4586–4591.
- Faith, J.J.; Guruge, J.L.; Charbonneau, M.; Subramanian, S.; Seedorf, H.; Goodman, A.L.; Clemente, J.C.; Knight, R.; Heath, A.C.; Leibel, R.L.; et al. The Long-Term Stability of the Human Gut Microbiota. Science 2013, 341, 1237439.
- Mayer, E.A. Gut feelings: The emerging biology of gut–brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466.
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712.
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front. Endocrinol. 2020, 11, 25.
- Gupta, A.; Osadchiy, V.; Mayer, E.A. Brain-gut-microbiome interactions in obesity and food addiction. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 655–672.
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158.
- Luczynski, P.; McVey Neufeld, K.-A.; Oriach, C.S.; Clarke, G.; Dinan, T.G.; Cryan, J.F. Growing up in a Bubble: Using Germ-Free Animals to Assess the Influence of the Gut Microbiota on Brain and Behavior. Int. J. Neuropsychopharmacol. 2016, 19, pyw020.
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.-N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275.
- Desbonnet, L.; Garrett, L.; Clarke, G.; Kiely, B.; Cryan, J.F.; Dinan, T.G. Effects of the probiotic Bifidobacterium infantis in the maternal separation model of depression. Neuroscience 2010, 170, 1179–1188.
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol. Motil. 2011, 23, 255–264.e119.
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; McCoy, K.D.; et al. The Intestinal Microbiota Affect Central Levels of Brain-Derived Neurotropic Factor and Behavior in Mice. Gastroenterology 2011, 141, 599–609.e3.
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; Dinan, T.G.; Cryan, J.F. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol. Psychiatry 2013, 18, 666–673.
- Schéle, E.; Grahnemo, L.; Anesten, F.; Hallén, A.; Bäckhed, F.; Jansson, J.-O. The Gut Microbiota Reduces Leptin Sensitivity and the Expression of the Obesity-Suppressing Neuropeptides Proglucagon (Gcg) and Brain-Derived Neurotrophic Factor (Bdnf) in the Central Nervous System. Endocrinology 2013, 154, 3643–3651.
- Chidambaram, S.B.; Rathipriya, A.G.; Mahalakshmi, A.M.; Sharma, S.; Hediyal, T.A.; Ray, B.; Sunanda, T.; Rungratanawanich, W.; Kashyap, R.S.; Qoronfleh, M.W.; et al. The Influence of Gut Dysbiosis in the Pathogenesis and Management of Ischemic Stroke. Cells 2022, 11, 1239.
- Chen, R.; Xu, Y.; Wu, P.; Zhou, H.; Lasanajak, Y.; Fang, Y.; Tang, L.; Ye, L.; Li, X.; Cai, Z.; et al. Transplantation of fecal microbiota rich in short chain fatty acids and butyric acid treat cerebral ischemic stroke by regulating gut microbiota. Pharmacol. Res. 2019, 148, 104403.
- Di Meo, F.; Donato, S.; di Pardo, A.; Maglione, V.; Filosa, S.; Crispi, S. New Therapeutic Drugs from Bioactive Natural Molecules: The Role of Gut Microbiota Metabolism in Neurodegenerative Diseases. Curr. Drug Metab. 2018, 19, 478–489.
- Farràs, M.; Martinez-Gili, L.; Portune, K.; Arranz, S.; Frost, G.; Tondo, M.; Blanco-Vaca, F. Modulation of the Gut Microbiota by Olive Oil Phenolic Compounds: Implications for Lipid Metabolism, Immune System, and Obesity. Nutrients 2020, 12, 2200.
- Varesi, A.; Campagnoli, L.I.M.; Fahmideh, F.; Pierella, E.; Romeo, M.; Ricevuti, G.; Nicoletta, M.; Chirumbolo, S.; Pascale, A. The Interplay between Gut Microbiota and Parkinson’s Disease: Implications on Diagnosis and Treatment. Int. J. Mol. Sci. 2022, 23, 12289.
- Memmola, R.; Petrillo, A.; di Lorenzo, S.; Altuna, S.C.; Habeeb, B.S.; Soggiu, A.; Bonizzi, L.; Garrone, O.; Ghidini, M. Correlation between Olive Oil Intake and Gut Microbiota in Colorectal Cancer Prevention. Nutrients 2022, 14, 3749.
- Ma, G.; Chen, Y. Polyphenol supplementation benefits human health via gut microbiota: A systematic review via meta-analysis. J. Funct. Foods 2020, 66, 103829.
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422.
- Rajha, H.N.; Paule, A.; Aragonès, G.; Barbosa, M.; Caddeo, C.; Debs, E.; Dinkova, R.; Eckert, G.P.; Fontana, A.; Gebrayel, P.; et al. Recent Advances in Research on Polyphenols: Effects on Microbiota, Metabolism, and Health. Mol. Nutr. Food Res. 2022, 66, 2100670.
- Corona, G.; Tzounis, X.; Dessì, M.A.; Deiana, M.; Debnam, E.S.; Visioli, F.; Spencer, J.P.E. The fate of olive oil polyphenols in the gastrointestinal tract: Implications of gastric and colonic microflora-dependent biotransformation. Free. Radic. Res. 2006, 40, 647–658.
- Santos, M.M.; Piccirillo, C.; Castro, P.; Kalogerakis, N.; Pintado, M.E. Bioconversion of oleuropein to hydroxytyrosol by lactic acid bacteria. World J. Microbiol. Biotechnol. 2012, 28, 2435–2440.
- Liu, Z.; Wang, N.; Ma, Y.; Wen, D. Hydroxytyrosol Improves Obesity and Insulin Resistance by Modulating Gut Microbiota in High-Fat Diet-Induced Obese Mice. Front. Microbiol. 2019, 10, 390.
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503.
- Li, H.; Christman, L.M.; Li, R.; Gu, L. Synergic interactions between polyphenols and gut microbiota in mitigating inflammatory bowel diseases. Food Funct. 2020, 11, 4878–4891.
- Thielmann, J.; Kohnen, S.; Hauser, C. Antimicrobial activity of Olea europaea Linné extracts and their applicability as natural food preservative agents. Int. J. Food Microbiol. 2017, 251, 48–66.
- Guo, W.; Liu, J.; Sun, J.; Gong, Q.; Ma, H.; Kan, X.; Cao, Y.; Wang, J.; Fu, S. Butyrate alleviates oxidative stress by regulating NRF2 nuclear accumulation and H3K9/14 acetylation via GPR109A in bovine mammary epithelial cells and mammary glands. Free Radic. Biol. Med. 2020, 152, 728–742.
- Wang, M.; Zhang, S.; Zhong, R.; Wan, F.; Chen, L.; Liu, L.; Yi, B.; Zhang, H. Olive Fruit Extracts Supplement Improve Antioxidant Capacity via AlteringColonic Microbiota Composition in Mice. Front. Nutr. 2021, 8, 645099.
- Gaupp, R.; Eledala, N.; Somerville, G.A. Staphylococcal response to oxidative stress. Front. Cell. Infect. Microbiol. 2012, 2, 33.
- Wang, N.; Ma, Y.; Liu, Z.; Liu, L.; Yang, K.; Wei, Y.; Liu, Y.; Chen, X.; Sun, X.; Wen, D. Corrigendum to “Hydroxytyrosol prevents PM2.5-induced adiposity and insulin resistance by restraining oxidative stress related NF-κB pathway and modulation of gut microbiota in a murine model” . Free. Radic. Biol. Med. 2022, 179, 426.
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436.
- Zheng, S.; Wang, Y.; Fang, J.; Geng, R.; Li, M.; Zhao, Y.; Kang, S.-G.; Huang, K.; Tong, T. Oleuropein Ameliorates Advanced Stage of Type 2 Diabetes in db/db Mice by Regulating Gut Microbiota. Nutrients 2021, 13, 2131.