Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Chaojun Ren and Version 2 by Rita Xu.
A stable life support system in the spacecraft can greatly promote long-duration, far-distance, and multicrew manned space flight. Therefore, controlling the concentration of CO
2
in the spacecraft is the main task in the regeneration system. The electrocatalytic CO
2
reduction can effectively treat the CO
2
generated by human metabolism. This technology has potential application value and good development prospect in the utilization of CO
2
in the space station.
- carbon dioxide
- electrocatalytic reduction
- electrocatalyst
- electrolyte
- reactor design
- spacecraft life support system
1. Introduction
With the continuous development of human science and technology, space exploration will face unprecedented opportunities and challenges [1][2][3][4][5][1,2,3,4,5]. Currently, realizing the recycle of materials in closed space stations is the best solution to solve the problem of water (H2O) and oxygen (O2) resupply faced by human activities, which not only can extend the duration of manned missions and reduce transportation costs, but also is a prerequisite for interstellar migration [6][7][8][9][10][6,7,8,9,10]. As the end product of human respiration and metabolic processes, carbon dioxide (CO2) carries a large amount of oxygen [11][12][13][11,12,13]. Therefore, recovering O2 from waste CO2 is an important way to achieve resource recycling and reduce transportation costs for medium- and long-term manned extraterrestrial missions [14][15][16][17][18][14,15,16,17,18].
The concentration of CO2 in the space station directly affects the life and health system of the astronauts [19]. To reduce the concentration of CO2 (<0.5%) and maintain the balance of air components, it is necessary to treat the CO2 emissions at 0.7–1.0 kg per person per day. How to better transform and recycle CO2 in the space station has become the focus of researchers [20][21][22][23][20,21,22,23]. The methods of CO2 transformation can be classified into two categories: physical absorption and chemical conversion [24][25][24,25]. The chemical conversion mainly includes thermochemistry, photocatalytic reduction, electrocatalytic reduction, and photoelectrocatalytic reduction [26][27][28][29][26,27,28,29]. Chemical conversion can capture and immobilize CO2 or convert it into useful low-carbon fuels, such as CO, CH4, HCOOH, CH3OH, etc. [30][31][32][33][34][30,31,32,33,34].
2. The Mechanism of Electrocatalytic Reduction of CO2
The electrocatalytic reduction of CO2 often occurs at the interface between the electrode and the electrolyte, and the electrolyte is usually an aqueous solution of potassium bicarbonate [19]. As shown in Figure 1, the multiphase catalytic process generally consists of four main steps: (i) The CO2 in the solution diffuses to the surface of the working electrode. (ii) The electrocatalyst adsorbs CO2 from the solution. (iii) Electron transfer or proton migration is used to cleave C-O bonds or form C-H bonds. (iv) The generated products are detached from the electrocatalyst surface and diffused into the electrolyte [35][36][60,61]. The adsorption of CO2 on the electrocatalyst surface is an important step in the whole reaction. During the adsorption process, the C=O bonding is strongly perturbed by the substrate, and the electrons are shared between the CO2 and the catalyst. A number of reaction mechanisms have been proposed for the conversion of CO2. It is widely accepted that the reactive species are the neutral hydrated CO2 molecules, and they will convert into CO2•− in the adsorption process on most of the main-group metal electrodes in aqueous media. Then the absorbed CO2•− reacts with the H2O molecules to form HCO2•. The intermediate would convert into to HCO2− because of its unstable unpaired electron, followed by the desorption of HCO2− species [35][60].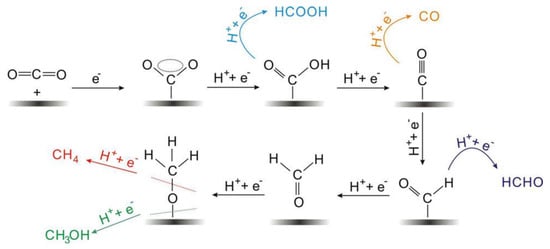
Figure 1. Reaction mechanism of electrochemical CO2 reduction on electrodes in aqueous solutions and the formation paths for the five main C1 products during the reduction process.
3. Electrocatalysts
Over the past few decades, researchers have attempted to develop high-performance CO2 electrocatalysts and have carried out extensive work [37][38][39][71,72,73]. The catalysts could reduce the activation energy required for the electroreduction of CO2, thereby minishing the reduction overpotential and current density. Most of the studies have focused on transition-metal-based catalysts, which are mainly Cu, Au, Fe, Ag, Re, Mn, Co, Ni, Pd, Ir, Ru, etc. [17][40][41][42][17,74,75,76]. The main forms of the complexes include transition metal polypyridines, metal porphyrins/phthalocyanines, and various metal phosphine complexes [43][44][77,78]. Among the transition metal complexes, Cu complexes had better activity. Recently, Raja et al. [44][78] designed a dinuclear Cu (I) complex as catalysts for the electroreduction of CO2, and its catalytic cycle is shown in Figure 2 below. They found that the Cu (I) system could be oxidized by CO2, which suggested that the selective bonding of CO2 and Cu (I) ions could provide a low-energy pathway for the formation of CO2•− radical anions. Thus, the Cu (II) tetranuclear oxalate-bridged complex [2]4+ was thermodynamically favorable. Moreover, the bonding of CO2 to the Cu(I) center in the dinuclear copper (I) complex [1]2+ was faster and has a higher selectivity. This was due to the low solubility of lithium oxalate in acetonitrile, and the release of the oxalate double anion from [2]4+ in the presence of LiClO4 could be accomplished instantaneously, and the dinuclear copper (II) complex [4]4+ was produced. Consequently, the electrocatalytic reduction of Cu (II) ions to Cu (I) might be the rate-controlling step in this system. The electrode surface was coated by the lithium oxalate generated during the reaction and then prevented the effective electron transfer.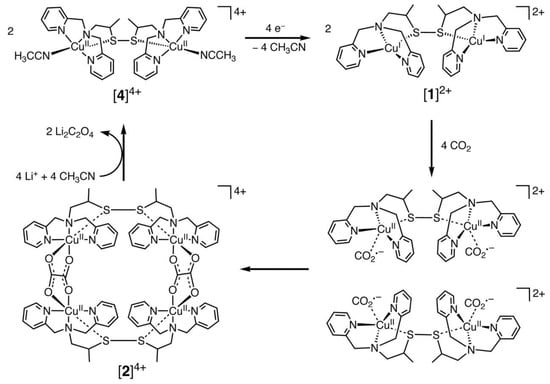
Figure 2. Proposed electrocatalytic cycle for oxalate formation.
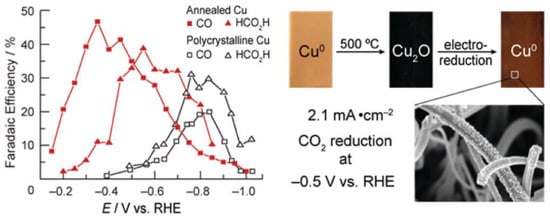
Figure 3. Electrocatalytic performance and the preparation process of Cu electrodes.
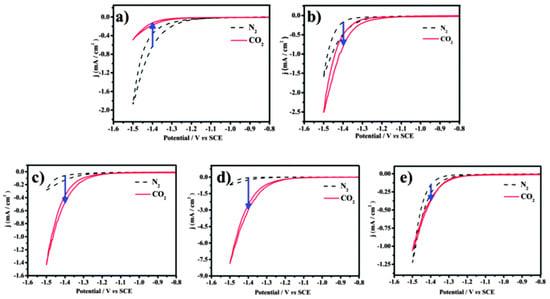
Figure 4. The cyclic voltammograms of catalysts were recorded in N2- and CO2-saturated 0.5 M KHCO3 with a scan rate of 10 mV/s−1 between −0.8 and −1.5 V (vs. SCE). (a) Cu-Pt-1#, (b) Cu-Pt-2#, (c) Cu-Pt-3#, (d) Cu-Pt-4#, and (e) Cu-Pt-5#.
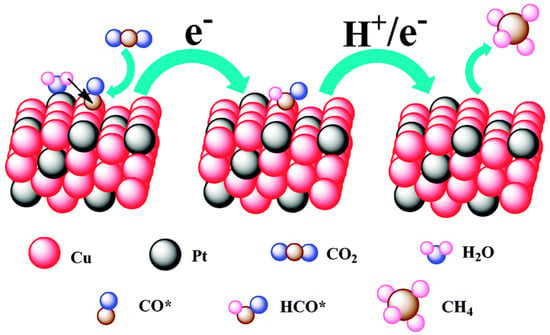
Figure 5. A proposed mechanism illustrating the steps of CO2 electroreduction and CH4 formation occurring at the Cu-Pt 3:1 NC catalyst.
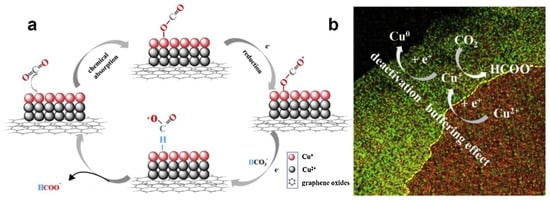
Figure 6. (a) A proposed mechanism illustrating the steps of electrochemical CO2 reduction and the formation of HCOOH occurring at the G-CuxO-2 h electrocatalyst. (b) A proposed “buffering effect” illustrating the origin of the durability of G-CuxO-T electrocatalysts.
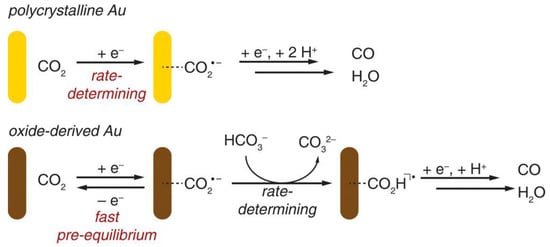
Figure 7. Proposed mechanisms for CO2 reduction to CO on polycrystalline Au and oxide-derived Au.
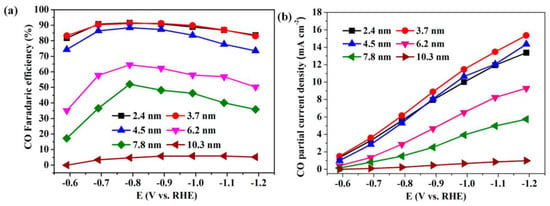
Figure 8. Applied potential dependence of (a) Faradaic efficiencies and (b) current densities for CO production over Pd NPs with different sizes.
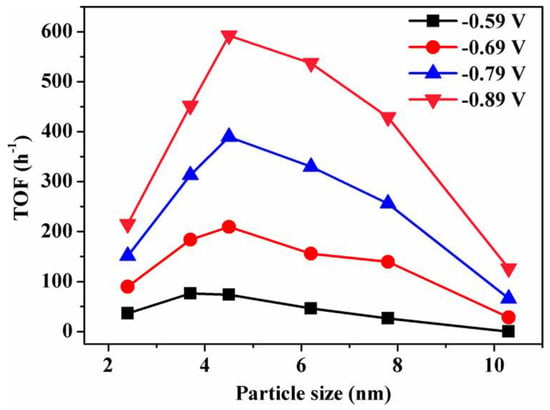
Figure 9. Size dependence of TOF for CO production on Pd NPs at various potentials.
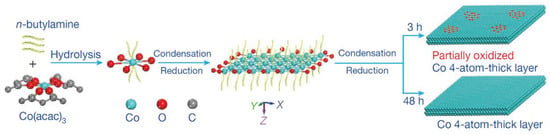
Figure 10. Schematic formation process of the partially oxidized and pure-Co 4-atomic-layer, respectively.
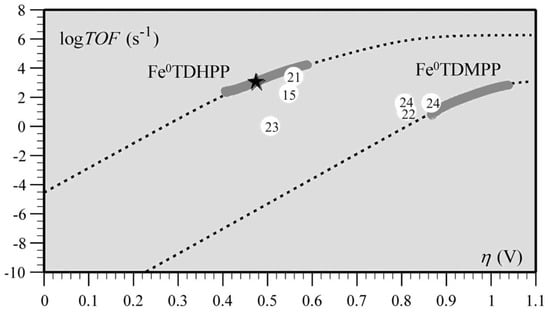
Figure 11. Correlation between turnover frequency and overpotential for the series of CO2-to-CO electroreduction catalysts. The star indicates TOF and η values from preparative-scale experiments of Fe0TDHPP.
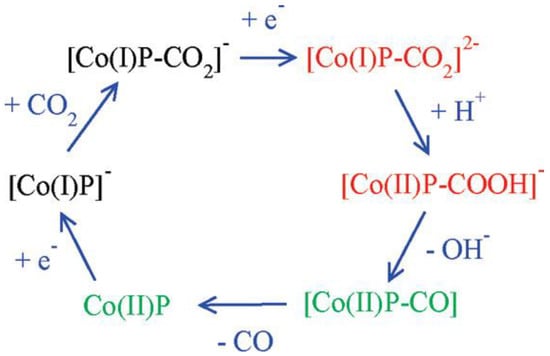
Figure 12. Mechanism of CO2 reduction with electron addition deduced from hybrid DFT plus dielectric continuum redox potential calculations. Red denotes key intermediates; green species should undergo fast reactions.
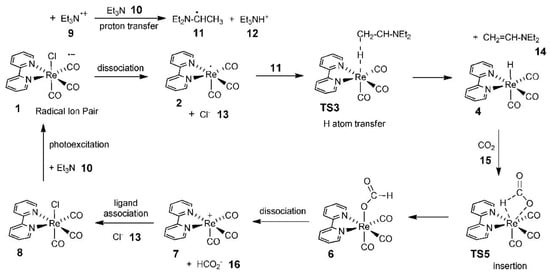
Figure 13. Computed photocatalytic cycle for CO2 reduction on a Re catalyst.