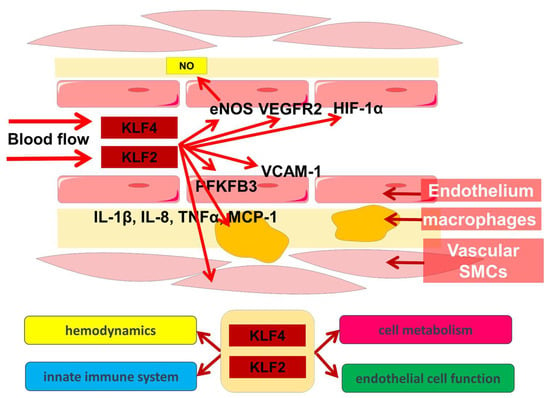
1. Regulation of Hemodynamics and Angiogenesis
Endothelial cells line the inner surface of blood vessels and are constantly exposed to hemodynamic forces arising from blood flow. The hemodynamic conditions to which endothelial cells are exposed can affect their structure and function. Shear stress is an important hemodynamic factor affecting endothelial function
[1][2][93,94]. Shear stress regulates the expression of a number of genes involved in endothelial function, including genes involved in inflammation, angiogenesis, and coagulation. In addition, shear stress also regulates the endothelial cell cytoskeleton. The cytoskeleton is represented by a dynamic network of protein filaments that provides structural support and plays a key role in cell migration, adhesion, and signal transduction. Shear stress has been shown to regulate the organization and dynamics of the cytoskeleton in endothelial cells
[3][4][95,96]. In response to shear stress, endothelial cells undergo significant cytoskeleton rearrangements, including reorientation and elongation of actin filaments
[5][6][97,98]. This contributes to the polarization of endothelial cells under shear stress. Polarization of endothelial cells involves the establishment of asymmetric distribution of organelles and proteins along the apical-basal axis of the cell
[7][8][99,100]. On the contrary, in the sections with the impaired flow, to which a low shear stress corresponds, the cells do not have a polarized shape. Such sections of arteries, which correspond to areas of curvatures and bifurcations, are more prone to atherogenesis
[2][9][94,101].
A growing body of evidence suggests cross-linkages between endothelial cell metabolism and function
[10][102]. Changes in glucose levels have been shown to affect the orientation of endothelial cells in the brain microcirculatory bed, including the orientation of cell nuclei and F-actin
[11][103]. The disintegration of the cytoskeletal structure of actin filaments in energy-depleted endothelial cells has been shown previously
[12][104]. Thus, glycolysis may be an important source of energy for cytoskeleton remodeling. The activity of glycolysis may be related to the regulation of the cytoskeleton, orientation, and polarization of endothelial cells. Inhibition of glycolysis in endothelial cells can disrupt apical-basal axis formation and disrupt actin cytoskeleton organization. It was shown that the glycolytic enzyme aldolase is involved in the formation of F-actin bundles through the stabilization of parallel F-actin filaments, which leads to the formation of a less organized branched F-actin cytoskeleton
[11][13][103,105]. At the same time, a higher level of glycolysis contributes to a greater involvement of aldolase in this process instead of F-actin stabilization, which leads to an increased migration ability of cells
[11][13][103,105]. In addition, the enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), which is a key regulator of glycolysis, plays a role in endothelial cell polarization and also promotes the expression of adhesion molecules such as Intercellular Adhesion Molecule 1 (ICAM-1), VCAM-1, and E-selectin
[14][15][106,107]. In addition, PFKFB3 increases VE-cadherin levels in the plasma membrane
[15][107]. PFKFB3 is also involved in endothelial cell migration and tube formation. Suppression of PFKFB3 impairs lamellipodia formation and impairs Tip cell activity, which is related to PFKFB3 participation in the control of filopodia/lamellipodia formation, which is partially mediated by its compartmentalization with F-actin in mobile protrusions
[16][108].
As noted above, KLF2 is activated by laminar shear stress and regulates a wide range of endothelial cell functions, including proliferation, migration, and inflammation
[17][18][46,109]. Overall, KLF2 is involved in the regulation of vascular hemodynamic forces and has a protective effect on the endothelium
[18][19][109,110]. While KLF2 is expressed at high levels in endothelial cells in the area of laminar flow, its expression drops to low levels near the sites of blood flow disturbances, which are characterized by the development of atherosclerosis
[17][46].
KLF2 overexpression was shown to reproduce the inhibitory effect of laminar flow on endothelial glycolysis. Shear stress acting through KLF2 reduced the expression of key glycolytic enzymes such as PFKFB3
[20][111]. KLF2 has been shown to suppress PFKFB3 promoter activity so that shear stress-mediated repression of endothelial cell metabolism controls their phenotype
[20][111]. Thus, laminar shear stress reduces PFKFB3 expression, which leads to a decrease in glycolysis. On the other hand, low shear stress and oscillatory flow can promote endothelial dysfunction, promoting a switch to glycolytic metabolism. High glucose levels are also relevant. Reduced KLF2 expression may exacerbate endothelial damage in diabetic nephropathy. In cell culture, it has been shown that KLF2 expression was suppressed with increasing glucose levels, whereas insulin, in contrast, led to increased KLF2 expression
[19][110]. At the same time, partial and temporary blockade of PFKFB3 leads to a decrease in glycolysis and reduces pathological angiogenesis
[21][112].
In addition, KLF2 has been shown to regulate the function of several cytoskeletal proteins. KLF2 regulates the phosphorylation and activity of cytoskeleton-related F-actin proteins
[22][113]. Shear stress promotes actin cytoskeleton remodeling through activation of the MEK5/ERK5/MEF2 MAPK pathway, which transcriptionally induces KLF2. KLF2 indirectly activates RhoA, which induces the formation of actin shear fibers that are necessary for both alignment in the flow direction and inhibition of the FAK-JNK-c-Jun/ATF2 signaling pathway
[22][113]. Thus, KLF2 promotes cell alignment in the flow direction through the formation of a thick network of actin shear fibers that is very different from classical tension fibers
[22][113].
KLF2 may also affect hemodynamics because it plays an important role in the stress-induced shear regulation of endothelial nitric oxide synthase (eNOS)
[23][114]. eNOS is an important regulator of hemodynamics through the production of nitric oxide (NO), which is considered a key player in the regulation of vascular tone through its vasorelaxant capacity. Reduced bioavailability of eNOS-derived NO is considered as a critical step in endothelial dysfunction and atherogenesis. Studies have shown that KLF2 induces eNOS expression and increases NO production in cultured human umbilical vein endothelial cells (HUVECs)
[24][115]. It is assumed that KLF2 promotes NO production by regulating eNOS uncoupling via Nrf2 (Nuclear factor erythroid 2-related factor 2)/ HO-1 (heme oxygenase-1)
[24][115]. Thus, activation of KLF2 in endothelial cells causes induction of eNOS and has a vasodilatory effect.
In turn, NRF2 regulates glycolysis and cell proliferation by regulating transcription of KLF2, PFKFB3, VEGFA, forkhead box protein O1 (FOXO1), and MYC, thereby controlling the switch between the active and resting states of the endothelium
[25][116]. This is because NRF2 is a regulator of miR-93 expression that targets both KLF2 and PFKFB3, which act as inhibitors and stimulators of glycolysis, respectively. This microRNA also acts on FOXO1, VEGFA, and MYC, which regulate cell proliferation
[16][20][25][26][108,111,116,117]. NRF2 has also been shown to be a known mediator of oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (oxPAPC) effects, which are produced by exposure of active oxygen species to PAPC, which is a component of cell membranes
[25][116]. It was shown that oxPAPC NRF2-dependently induces glycolysis and proliferation in endothelial cells. At the same time, endothelial cells
[25][116] are a significant source of extracellular miR-93, which are found in the exosomal fraction
[25][116].
KLF2 has a potent anti-angiogenic effect. KLF2 has been shown to induce the expression of genes that promote cell cycle arrest and inhibit cell proliferation, presumably by affecting the VEGF-mediated pathway. The molecular mechanism is the ability of KLF2 to inhibit VEGFR2/KDR expression
[27][118]. KLF2 also attenuates cell migration by affecting multiple genes, including VEGFR2 and SEMA3F (Semaphorin 3F)
[23][114]. Thus, when KLF2 levels in cells are increased, VEGFR2 levels are decreased
[23][114]. In general, KLF2 helps to maintain the integrity of the endothelial monolayer and prevent the formation of atherosclerotic lesions
[28][32].
KLF2 has also been shown to affect angiogenesis through the inhibition of hypoxia-inducible factor 1-alpha (HIF-1α) by promoting HIF-1α degradation, affecting its folding and maturation
[29][119]. KLF2 overexpression was shown to significantly inhibit hypoxia-induced endothelial tube formation, whereas KLF2 deficiency promotes hypoxia-mediated angiogenesis in vivo
[29][119].
Another member of the KLF family, KLF4, has many similar functions as KLF2. KLF4 plays a key role in the regulation of the endothelial cell cytoskeleton. KLF4 has been shown to regulate the function of several cytoskeletal proteins
[30][120]. In addition to the regulation of cytoskeletal proteins, KLF4 also regulates the expression of proteins involved in the formation and maintenance of endothelial cell junctions. Endothelial cell junctions are crucial for maintaining endothelial integrity and regulating vascular tone and blood flow. KLF4 has been shown to regulate the expression of several functional proteins, including VE-cadherin
[31][121]. At the same time, Klf4 knockdown disrupted the endothelial barrier and also enhanced lipopolysaccharide-induced lung injury and pulmonary edema in mice
[31][121].
In addition, KLF4 is also involved in other hemodynamic mechanisms, including eNOS regulation
[32][33][122,123]. At the same time, vasorelaxation in pulmonary arteries has been shown to be impaired under nitrosative stress, which is associated with S-nitrosation of KLF4. At the same time, endothelin-1 stimulated S-nitrosylation of KLF4 in endothelial cells
[33][123]. These findings are of interest because of the role of smoking in the pathogenesis of atherosclerosis and chronic obstructive pulmonary disease and the role of nitrosative stress in their development.
Thus, KLF2 and KLF4 are important participants in hemodynamic regulation, and this function has close cross-linkages with other endothelial cell functions.
2. Immunometabolism of Endothelial Cells
In addition to the fact that endothelial cells play an important role in vascular functions such as vasodilation, blood pressure regulation, and angiogenesis, a growing body of evidence is increasing the understanding that endothelial cells are also involved in immune responses
[34][35][124,125]. Endothelial cell function also includes the regulation of immune cell migration across the vascular barrier through the production of immune mediators such as cytokines and chemokines. Endothelial cells express several classes of molecules, such as selectins and integrins, which are involved in leukocyte adhesion and extravasation from the bloodstream. Endothelial cells may also themselves undergo an inflammatory response with activation of adhesion molecules and secretion of proinflammatory cytokines in response to stimuli such as LPS or other pathogens
[35][36][125,126]. Endothelial cell activation is accompanied by a change in their metabolism, which is known as cellular immunometabolism. The significance of cellular immunometabolism is well known in the example of macrophage polarization. Macrophages are known to exhibit a differentiated role in inflammation. In addition to pro-inflammatory, “classically activated” M1 macrophages, “alternatively activated” M2 macrophages are known to contribute to the resolution of inflammation. Switching cellular metabolism to glycolysis is an important step in the M1 activation of macrophages.
Glycolysis is the main source of energy for endothelial cells
[10][37][38][39][102,127,128,129]. Although endothelial cells have good access to oxygen from the blood, these cells produce up to 85% of ATP as a result of glycolysis rather than oxidative phosphorylation. Thus, under conditions of migration, proliferation, or stress, endothelial cells increase glycolysis to meet energy requirements
[39][40][41][129,130,131]. Increased glycolysis plays an important role in the immunometabolism of endothelial cells, providing them with energy and metabolites for the immune response.
High levels of glycolysis in endothelial cells are associated with the regulation of several rate-limiting stages, which are related to the action of hexokinase 2 (HK2) and phosphofructokinase 1 (PFK1). Meanwhile, PFK1 activity is controlled by PFKFB3, which produces fructose-2,6-bisphosphate, the main allosteric activator of PFK1
[42][43][132,133]. Glycolysis in endothelial cells is regulated by various signaling pathways, including the PI3K/Akt and MAPK pathways
[44][134]. These pathways are activated in response to proinflammatory signals that activate glycolytic enzymes such as hexokinase and phosphofructokinase.
PFKFB3 plays an important role in the regulation of glycolysis, especially under pathological conditions and during angiogenesis
[16][21][45][46][108,112,135,136]. In an experiment with primary human pulmonary artery endothelial cells and aortic endothelial cells that were stimulated with TNFα or LPS, or TNFα and LPS were shown to increase glucose uptake 2.2-fold, glycolysis 2.3-fold, and mitochondrial respiration 1.6-fold. The increase in glycolysis correlates with a 2-4-fold activation of PFKFB3
[20][47][111,137]. At the same time, TNFα and LPS were shown to stimulate the expression of mRNA and the KLF4 protein, which acts as a repressor of PFKFB3 transcription. KLF4 was also found to suppress inflammation by normalizing PFKFB3-mediated enhancement of glycolysis, and the KLF4-PFKFB3 signaling axis integrates immunometabolism in human arterial endothelial cells
[20][47][111,137].
In addition, glycolysis is also regulated by transcription factors such as HIF-1α and c-Myc
[48][49][138,139]. HIF-1α is activated under conditions of hypoxia, which is characteristic of the inflammatory environment and activates glycolytic enzymes, promoting ATP production
[44][134]. Reduced c-Myc expression in endothelial cells leads to a proinflammatory senescent phenotype characterized by an enhanced inflammatory response and endothelial dysfunction
[50][140].
KLF2 also modulates the inflammatory response in atherosclerosis
[51][141]. KLF2 has been shown to suppress the expression of proinflammatory genes such as VCAM-1 and MCP-1 and promote the expression of anti-inflammatory genes such as eNOS and HO-1
[23][51][52][53][54][55][56][57][34,114,141,142,143,144,145,146]. This anti-inflammatory effect helps prevent the migration of monocytes and other inflammatory cells to the vessel wall and limits the development of atherosclerotic lesions.
Thus, KLF2 plays an important role in the regulation of proinflammatory activation of both endothelial cells and monocytes
[58][31]. The anti-inflammatory effect of KLF2 has been shown to be related to the inhibition of monocyte proinflammatory gene expression, which inhibited the expression of several cytokines/chemokines such as IL-1β, IL-8, TNFα, MCP-1, and inflammatory factors, including tissue factor and COX-2
[56][145]. KLF2 exerts its anti-inflammatory effect through a decrease in the pro-inflammatory activity of NF-κB
[56][145]. On the other hand, NF-κB inhibits KLF2 expression by interrupting the binding of MADS box transcription enhancer factor 2 (MEF2) and histone deacetylase (HDAC) molecule access to the KLF2 promoter
[58][31]. Thus, KLF2 and NF-κB interact to regulate inflammatory pathways
[58][31]. Interestingly, however, the consumption of fatty foods decreases myeloid KLF2 levels, which represents a mechanism for cross-linking risk factors in atherogenesis
[59][147].
The role of KLF4 in atherosclerosis is complex and cell-type-dependent. KLF4 may play a protective role in endothelium but a pathogenic role in smooth muscle cells. Overexpression of both Klf4 and KLF2 has been shown to increase the expression of several anti-inflammatory and antithrombotic factors in endothelial cells
[60][45]. Klf4 also inhibits TNF-α-induced expression of Vcam1 by blocking the binding of nuclear factor-κB to the Vcam1 promoter
[61][148]. Interestingly, statins increase KLF4 expression, with KLF4 mediating the suppressive effect of statins on TNF-α-induced VCAM1 expression by reducing NF-κB binding to the VCAM1 promoter
[62][149].
Meanwhile, KLF4 plays a key role in the proliferation and differentiation of VSMCs, acting as a “molecular switch” of their function
[63][52]. Phenotypic switching and proliferation of VSMCs is an important part of atherogenesis. It was previously shown that KLF4 knockout mice exhibit enhanced neointima formation compared to control mice
[64][150]. In this case, KLF4 acts as a suppressor of VSMCs differentiation and also as an inhibitor of proliferation of these cells in response to vascular injury
[63][52]. Klf4 suppresses the proliferation of smooth muscle cells as well as the expression of numerous markers of their differentiation, such as smooth muscle (SM) 22α and SM α-actin
[61][65][148,151]. On the other hand, cholesterol-loaded smooth muscle cells can induce KLF4 expression and transform smooth muscle cells into a foam cell phenotype, which is a key feature of atherosclerosis
[66][152].
KLF4 has an anti-inflammatory effect on myeloid cells, whereas KLF4 deficiency in myeloid tissue leads to increased inflammatory cellular infiltration of atherosclerotic lesions
[67][153]. KLF4 overexpression in macrophages has been shown to promote an anti-inflammatory M2 phenotype through transcriptional interactions with STAT6, whereas KLF4 deficiency in macrophages promotes a proinflammatory M1 phenotype
[68][69][70][71][39,154,155,156].
Thus, metabolic and immune processes are closely cross-linked, with KLF2 and KLF4 playing an important role in their regulation (Figure 1).
3. Participation of KLF2 and KLF4 in Other Biological Processes in the Vascular Wall
Thus, KLF2 has a role in protecting the endothelium from inflammation and damaging factors, which plays an important role in preventing atherosclerosis
[72][73][157,158]. In addition, KLF2 has been shown to regulate the expression of genes involved in lipid metabolism and transport, which are important for the development of atherosclerosis
[74][159]. KLF2 has been shown to promote the expression of genes involved in cholesterol efflux, such as ATP binding cassette subfamily A member 1 (ABCA1) and ATP binding cassette subfamily G member 1 (ABCG1), which help remove excess cholesterol from macrophages and prevent the formation of foam cells, a hallmark of atherosclerosis. In addition, KLF2 deficiency is involved in the formation of primary macrophage foam cells through the potential regulation of the key protein FABP4 (fatty-acid binding protein 4), which transports fatty acids and is expressed in adipocytes and macrophages
[57][146]. KLF2 has been shown to inhibit monocyte activation and phagocytic capacity
[56][145]. At the same time, KLF2 expression in circulating monocytes is reduced in patients with coronary heart disease
[56][145]. This is consistent with the evidence of increased lipid uptake and aP2/ FABP4 expression in KLF2-deficient hemizygous macrophages. Increased lipid uptake by macrophages may be responsible for increased atherosclerosis
[57][146]. The results of these studies suggest that increasing KLF2 expression may be a novel strategy for preventing and treating atherosclerosis
[75][160].
KLF4 increases the levels of cholesterol-25-hydroxylase and liver receptor X in endothelial cells and macrophages, which promotes an atheroprotective synergistic effect between these cells. KLF4-mediated transactivation of cholesterol-25-hydroxylase and hepatic X receptor promotes cholesterol efflux and M1-to-M2 conversion in macrophages
[76][161]. These data reinforce the understanding that KLF2 and KLF4 have functional overlaps in myeloid and endothelial cells, demonstrating a similar evolutionary trajectory
[68][39].
Of interest are the roles of microRNAs in KLF2 and KLF4 function and the overlap between lipid metabolism and hemodynamics. MiR-92a is involved in the regulation of KLF2 and KLF4 in endothelial cells
[23][52][60][77][78][34,45,114,162,163]. The expression of miR-92a is increased in endothelial cells and blood flow in atherosclerosis. It has been shown that in turbulent blood flow, there is an increase in miR-92a expression, while in laminar flow, there is a decrease. Inhibition of miR-92a prevents endothelial dysfunction and atherosclerosis in mice
[79][164].
This is because, acting through KLF2 and KLF4, miR-92a regulates endothelial cell activation by oxLDL under low-shear stress conditions
[79][164]. Inhibition of miR-92a was shown to reduce ICAM-1 expression when exposed to low shear voltage both in the presence and absence of oxLDL. At the same time, miR-92a overexpression caused a significant increase in ICAM-1 expression
[79][164].
In addition, miR-92a controls angiogenesis and functional repair of ischemic tissues by acting through integrin Subunit Alpha 5 (ITGA5) mRNA
[80][165]. Downregulation of eNOS in response to miR-92a overexpression has also been shown to occur secondary to ITGA5 mRNA degradation
[80][165].
Importantly, endothelial miR-92a is transported into macrophages mainly via extracellular vesicles, where it contributes to macrophage inflammatory activation
[81][166]. miR-92a can also bind to high-density lipoprotein (HDL) in the serum of patients with coronary heart disease (CHD)
[82][167]. In patients with CHD, miR-92a levels were significantly elevated, with endothelial cells being the main source of cells for miR-92-containing microvesicles. miR-92a transport by microvesicles is considered to be an important mechanism of angiogenesis regulation and a tool of intercellular communication
[83][168]. Transported into macrophages mainly through extracellular vesicles, miR-92a regulates KLF4 levels, contributing to their atherogenic phenotypic switching, inflammatory activation, and increased LDL uptake
[81][166]. ABCA1 is a direct target of miR-92a and is a component of the miR-17-92 cluster
[84][169]. ABCA1 is an important participant in reverse cholesterol transport, a process by which cellular cholesterol is exported to an extracellular acceptor with the formation of LDL. Importantly, excessive cholesterol accumulation due to reduced expression or functional activity of ABCA1 leads to their proinflammatory activation through several mechanisms. These data enhance the understanding of the role of miR-92a as an important participant in providing various mechanisms of arterial homeostasis
[81][166].
Endothelial-mesenchymal transition (EndMT) is a process of cell differentiation in which endothelial cells acquire mesenchymal properties. EndMT is associated with the development of atherosclerosis and can cause a number of phenotypic changes in endothelial cells, including their dysfunction as well as plaque formation
[85][170]. Unidirectional laminar flow protects endothelial cells from EndMT, whereas impaired flow promotes EndMT formation. Laminar flow has been shown to induce the expression of TN-X (tenascin-X) extracellular matrix protein in mouse and human endothelial cells via the transcription factor KLF4. In turn, TN-X inhibits the activity of TGF-β and EndMT
[86][171]. In addition, PFKFB3 is considered to be a critical factor of EndMT
[87][172]. These data enhance the understanding of the role of KLF4 in EndMT regulation. KLF2 has also been shown to inhibit TGF-beta signaling through the induction of inhibitory Smad7 and attenuation of AP-1 activity
[88][173].
KLF2 is also considered as a transcriptional regulator of endothelial thrombotic function
[89][174]. KLF2 overexpression increased clotting time as well as blood flow velocity in basal and inflammatory conditions. This is due to KLF2-mediated induction of thrombomodulin (TM) and eNOS expression and decreased expression of plasminogen activator inhibitor-1 (PAI-1)
[89][174]. KLF4 also protects against atherothrombosis in mice by inducing an anti-adhesive and anti-thrombotic state of endothelial cells
[90][175].
Laminar flow has also been shown to inhibit vascular calcification by inhibiting endothelial bone morphogenetic protein (BMP)/SMAD1/5 signaling via KLF2
[91][176]. Vascular calcification plays an important role in the pathogenesis of atherosclerosis; thus, the role of KLF2 is of clinical interest.
Thus, KLF2 and KLF4 play crucial roles in the molecular mechanisms of atherosclerosis, regulating multiple pathways involved in endothelial cell function and other vascular wall and blood flow cells, as well as in inflammation and lipid metabolism. The protective effects of KLF2 and KLF4 in atherosclerosis make them an attractive target for the development of new treatments for cardiovascular diseases.