The liver is a major store of glycogen and is essential in maintaining systemic glucose homeostasis. In healthy individuals, glycogen synthesis and breakdown in the liver are tightly regulated. Abnormal glycogen metabolism results in prominent pathological changes in the liver, often manifesting as hepatic glycogenosis or glycogen inclusions. This can occur in genetic glycogen storage disease or acquired conditions with insulin dysregulation such as diabetes mellitus and non-alcoholic fatty liver disease or medication effects.
Excessive glycogen accumulation within hepatocytes occurs in diseases caused or accompanied by the dysregulation of carbohydrate metabolism, as well as in conditions not primarily driven by altered carbohydrate metabolism. In these pathological conditions, the excess glycogen is distinctly visible on H&E; the hepatocytes usually exhibit cytoplasmic pallor and rarefaction or may show cytoplasmic-glycogen-filled inclusion bodies. Some hepatocytes may also demonstrate glycogen-filled nuclear vacuoles without a delimiting membrane (“glycogenated nuclei”); these are frequently seen in patients with diabetes and obesity, although they can also be seen in other liver conditions such as Wilson’s disease.
- glycogenic hepatopathy
- Mauriac syndrome
- hepatic glycogenosis
1. Glycogenic Hepatopathy
1.1. Clinical Findings and Pathogenesis
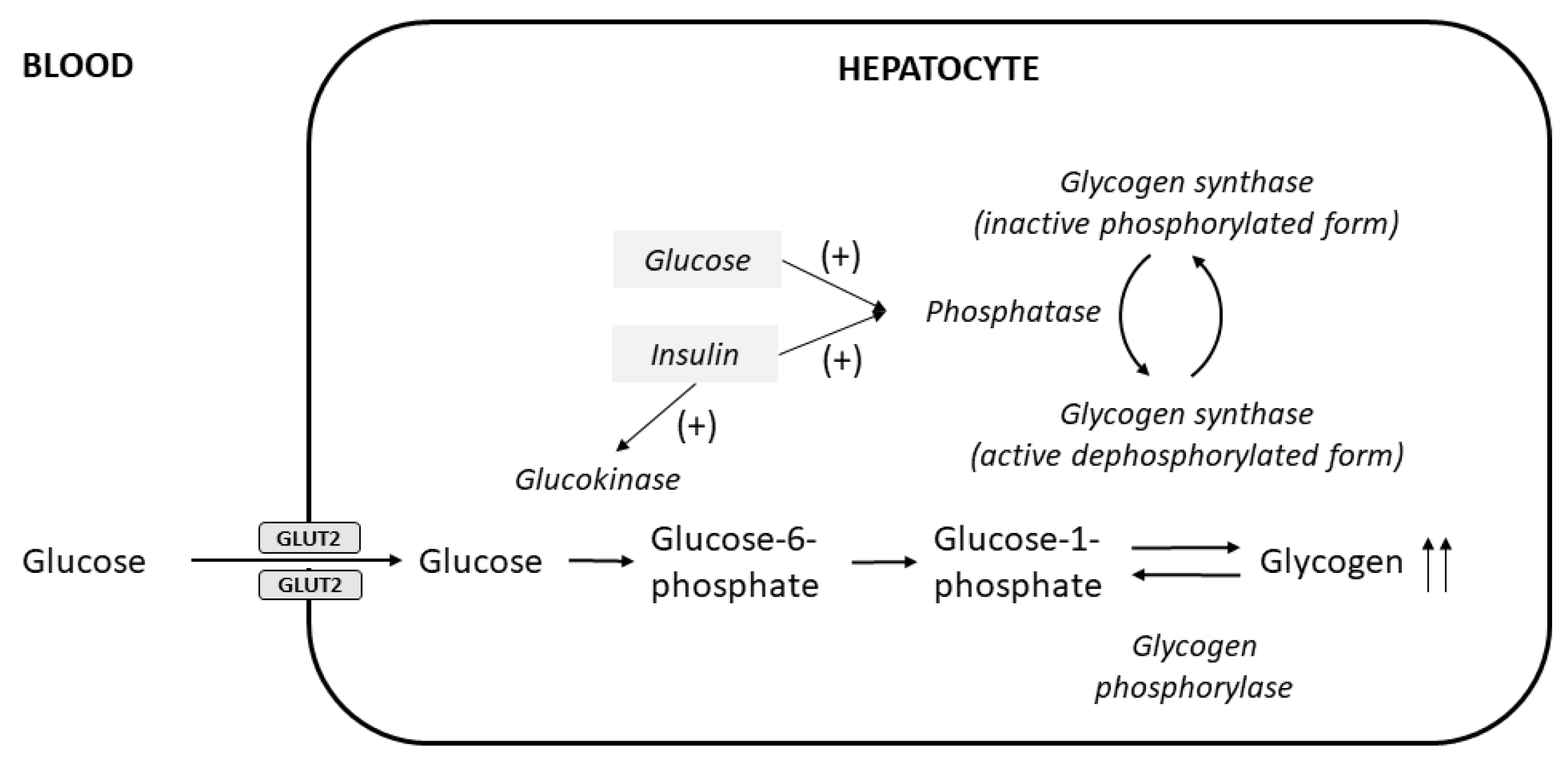
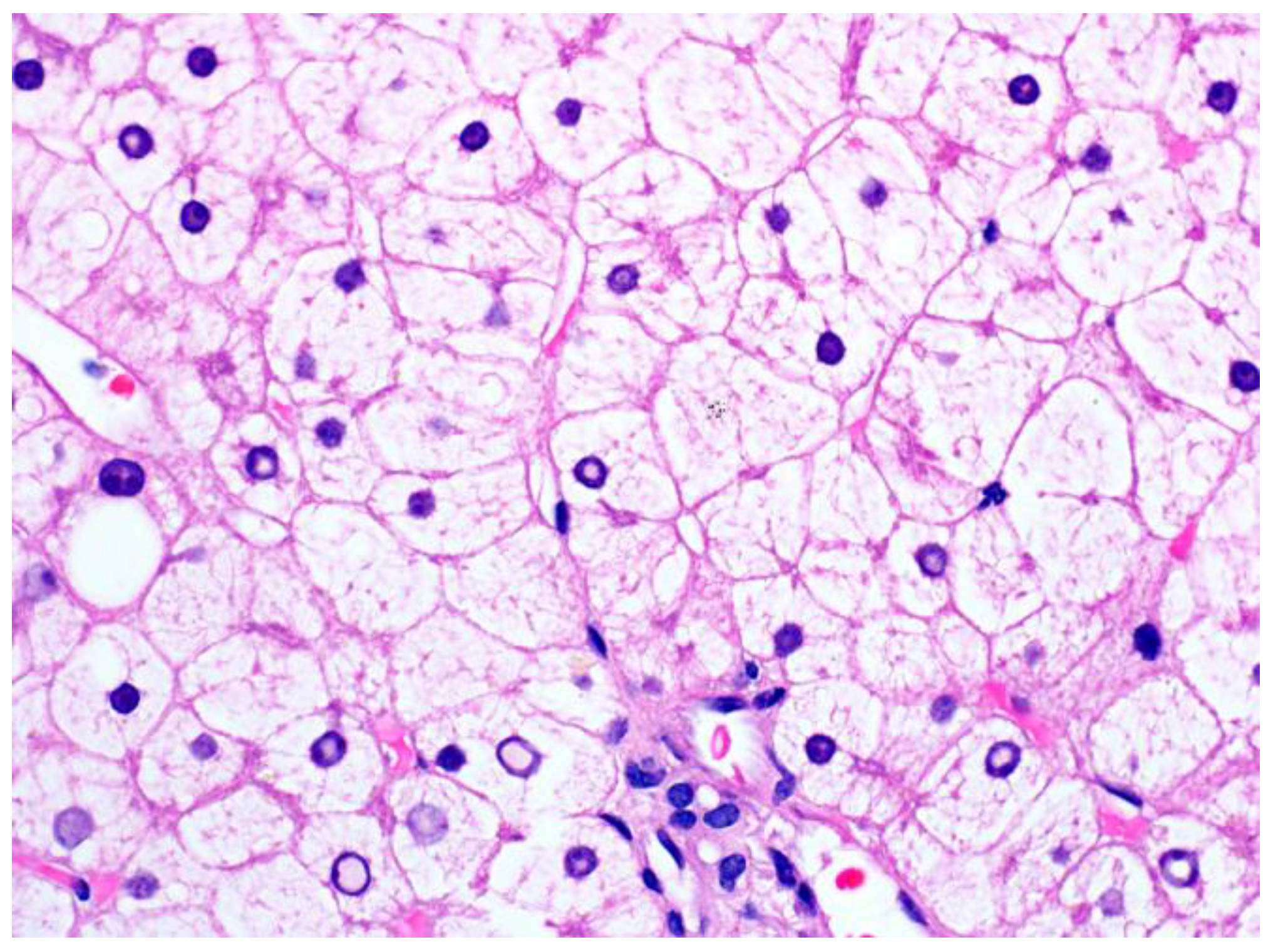
1.2. Pathological Findings
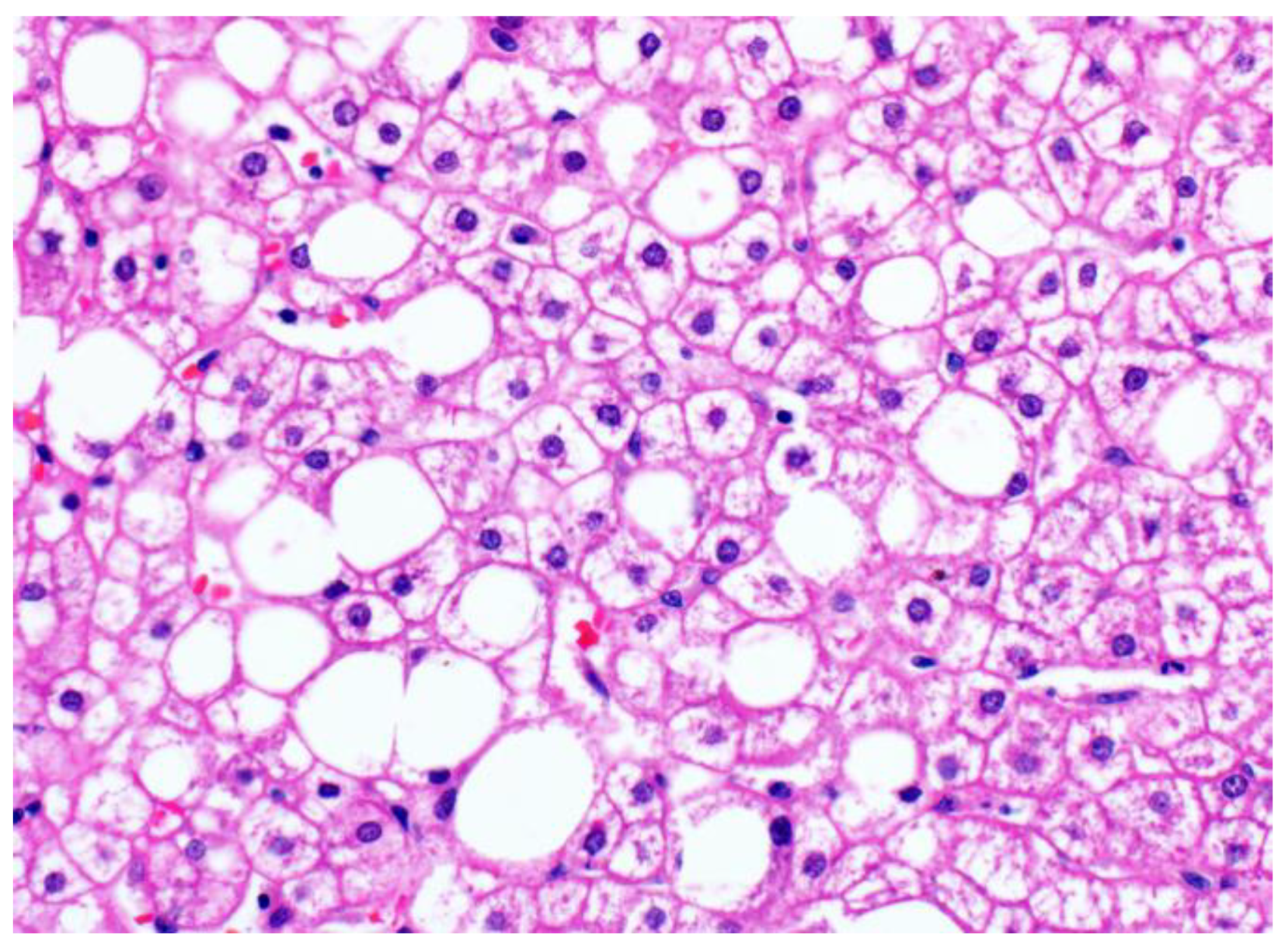
2. Glycogenosis in NAFLD
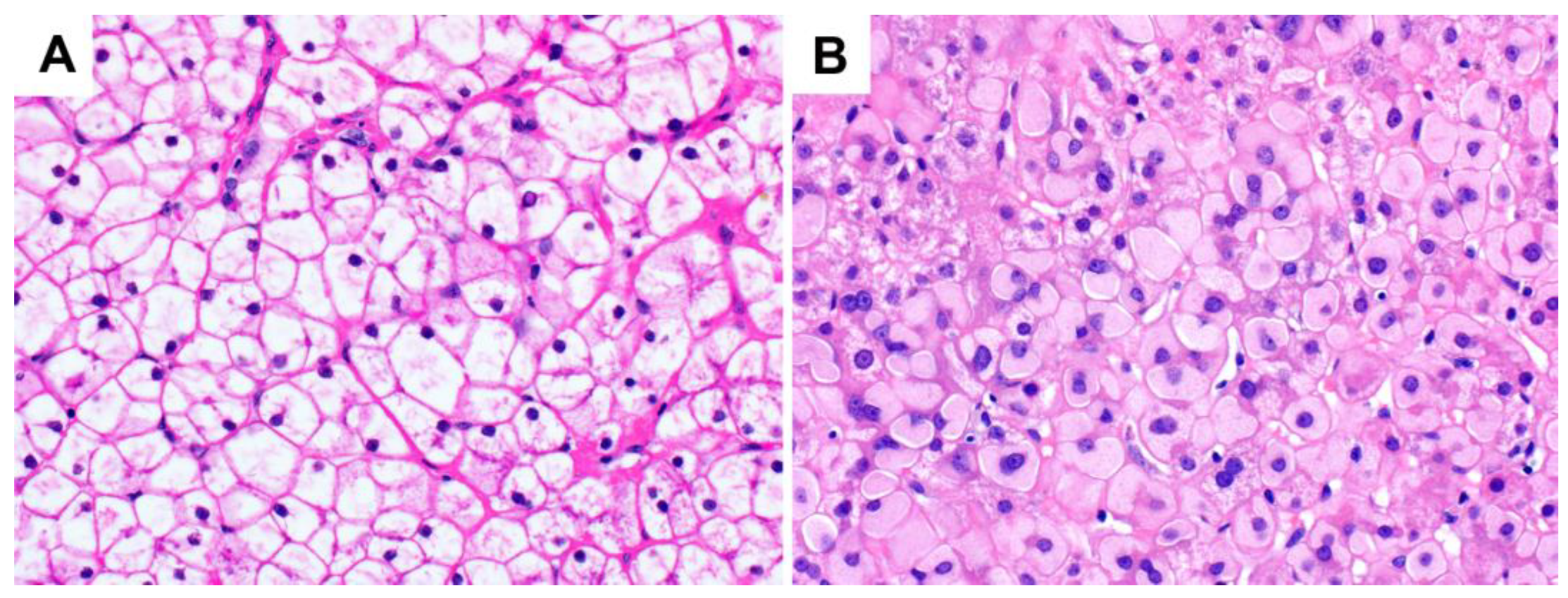
3. Glycogen Storage Disease
| Types | Gene Mutation | Defective Protein | Clinical Manifestations | Main Liver Findings |
|---|---|---|---|---|
| 0 | GYS2 | Glycogen synthetase | Hypoglycemia | Steatosis |
| Ia | G6PC | Glucose-6-phosphatase | Short stature, hepatomegaly, hypoglycemia, and increased triglycerides, lactate, and uric acid | Steatosis Glycogenosis |
| Ib | SLC37A4 | Glucose-6-phosphate translocase | Similar to type Ia with additional neutropenia, infections, and poor wound healing | Steatosis Glycogenosis |
| III | AGL | Debranching enzyme and variants | Short stature, hepatomegaly, hypoglycemia, hyperlipidemia, and cardiomyopathy | Steatosis Glycogenosis |
| IV | GBE1 | 1,4-alpha-glucan branching enzyme 1 | Progressive form: short stature, hepatomegaly, splenomegaly, cirrhosis, and late-onset hypoglycemia Non-progressive form: hepatomegaly |
Hepatocyte inclusions resembling ground glass changes |
| VI | PYGL | Liver phosphorylase | Short stature, hepatomegaly, and hypoglycemia | Steatosis Glycogenosis |
| IX | PHKA1, PHKA2, PHKB, PHKG2 | Phosphorylase kinase and variants | Short stature, hepatomegaly, hypoglycemia, hyperlipidemia, and myopathy | Glycogenosis |
References
- Mauriac, P. Gros Ventre, Hepatomegalie, Troubles de Las Croissance Chez Les Enfants Diabetiques Traits Depuis Plusieurs Annes Par l’insuline. Gax. Hebd. Med. Bordx. 1930, 26, 402–410.
- Carcione, L.; Lombardo, F.; Messina, M.F.; Rosano, M.; De Luca, F. Liver Glycogenosis as Early Manifestation in Type 1 Diabetes Mellitus. Diabetes Nutr. Metab. 2003, 16, 182–184.
- Chatila, R.; West, A.B. Hepatomegaly and Abnormal Liver Tests Due to Glycogenosis in Adults with Diabetes. Medicine 1996, 75, 327–333.
- Evans, R.W.; Littler, T.R.; Pemberton, H.S. Glycogen Storage in the Liver in Diabetes Mellitus. J. Clin. Pathol. 1955, 8, 110–113.
- Torres, M.; López, D. Liver Glycogen Storage Associated with Uncontrolled Type 1 Diabetes Mellitus. J. Hepatol. 2001, 35, 538.
- Nakamuta, M.; Ohashi, M.; Goto, K.; Tanabe, Y.; Hiroshige, K.; Nawata, H. Diabetes Mellitus-Associated Glycogen Storage Hepatomegaly: Report of a Case and Review of the Japanese Literature. Fukuoka Igaku Zasshi 1993, 84, 354–358.
- Torbenson, M.; Chen, Y.-Y.; Brunt, E.; Cummings, O.W.; Gottfried, M.; Jakate, S.; Liu, Y.-C.; Yeh, M.M.; Ferrell, L. Glycogenic Hepatopathy: An Underrecognized Hepatic Complication of Diabetes Mellitus. Am. J. Surg. Pathol. 2006, 30, 508–513.
- Haffar, S.; Izzy, M.; Habib, H.; Sugihara, T.; Li, D.K.; Sharma, A.; Wang, Z.; Murad, M.H.; Watt, K.D.; Bazerbachi, F. Liver Chemistries in Glycogenic Hepatopathy Associated with Type 1 Diabetes Mellitus: A Systematic Review and Pooled Analysis. Liver Int. 2021, 41, 1545–1555.
- Bronstein, H.D.; Kantrowitz, P.A.; Schaffner, F. Marked Enlargement of the Liver and Transient Ascites Associated with the Treatment of Diabetic Acidosis. N. Engl. J. Med. 1959, 261, 1314–1318.
- Mukewar, S.; Sharma, A.; Lackore, K.A.; Enders, F.T.; Torbenson, M.S.; Kamath, P.S.; Roberts, L.R.; Kudva, Y.C. Clinical, Biochemical, and Histopathology Features of Patients With Glycogenic Hepatopathy. Clin. Gastroenterol. Hepatol. 2017, 15, 927–933.
- Fitzpatrick, E.; Cotoi, C.; Quaglia, A.; Sakellariou, S.; Ford-Adams, M.E.; Hadzic, N. Hepatopathy of Mauriac Syndrome: A Retrospective Review from a Tertiary Liver Centre. Arch. Dis. Child. 2014, 99, 354–357.
- Munns, C.F.; McCrossin, R.B.; Thomsett, M.J.; Batch, J. Hepatic Glycogenosis: Reversible Hepatomegaly in Type 1 Diabetes. J. Paediatr. Child Health 2000, 36, 449–452.
- Tsujimoto, T.; Takano, M.; Nishiofuku, M.; Yoshiji, H.; Matsumura, Y.; Kuriyama, S.; Uemura, M.; Okamoto, S.; Fukui, H. Rapid Onset of Glycogen Storage Hepatomegaly in a Type-2 Diabetic Patient after a Massive Dose of Long-Acting Insulin and Large Doses of Glucose. Intern. Med. 2006, 45, 469–473.
- Umpaichitra, V. Unusual Glycogenic Hepatopathy Causing Abnormal Liver Enzymes in a Morbidly Obese Adolescent with Well-Controlled Type 2 Diabetes: Resolved after A1c Was Normalized by Metformin. Clin. Obes. 2016, 6, 281–284.
- Jiang, S.; Young, J.L.; Wang, K.; Qian, Y.; Cai, L. Diabetic-induced Alterations in Hepatic Glucose and Lipid Metabolism: The Role of Type 1 and Type 2 Diabetes Mellitus (Review). Mol. Med. Rep. 2020, 22, 603–611.
- Allende, D.S.; Gawrieh, S.; Cummings, O.W.; Belt, P.; Wilson, L.; Van Natta, M.; Behling, C.A.; Carpenter, D.; Gill, R.M.; Kleiner, D.E.; et al. Glycogenosis Is Common in Nonalcoholic Fatty Liver Disease and Is Independently Associated with Ballooning, but Lower Steatosis and Lower Fibrosis. Liver Int. 2021, 41, 996–1011.
- Petersen, M.C.; Vatner, D.F.; Shulman, G.I. Regulation of Hepatic Glucose Metabolism in Health and Disease. Nat. Rev. Endocrinol. 2017, 13, 572–587.
- Iancu, T.C.; Shiloh, H.; Dembo, L. Hepatomegaly Following Short-Term High-Dose Steroid Therapy. J. Pediatr. Gastroenterol. Nutr. 1986, 5, 41–46.
- Resnick, J.M.; Zador, I.; Fish, D.L. Dumping Syndrome, a Cause of Acquired Glycogenic Hepatopathy. Pediatr. Dev. Pathol. 2011, 14, 318–321.
- Kransdorf, L.N.; Millstine, D.; Smith, M.L.; Aqel, B.A. Hepatic Glycogen Deposition in a Patient with Anorexia Nervosa and Persistently Abnormal Transaminase Levels. Clin. Res. Hepatol. Gastroenterol. 2016, 40, e15–e18.
- Komuta, M.; Harada, M.; Ueno, T.; Uchimura, Y.; Inada, C.; Mitsuyama, K.; Sakisaka, S.; Sata, M.; Tanikawa, K. Unusual Accumulation of Glycogen in Liver Parenchymal Cells in a Patient with Anorexia Nervosa. Intern. Med. 1998, 37, 678–682.
- Manderson, W.G.; McKiddie, M.T.; Manners, D.J.; Stark, J.R. Liver Glycogen Accumulation in Unstable Diabetes. Diabetes 1968, 17, 13–16.
- MacDonald, M.J.; Hasan, N.M.; Ansari, I.-U.H.; Longacre, M.J.; Kendrick, M.A.; Stoker, S.W. Discovery of a Genetic Metabolic Cause for Mauriac Syndrome in Type 1 Diabetes. Diabetes 2016, 65, 2051–2059.
- Tomihira, M.; Kawasaki, E.; Nakajima, H.; Imamura, Y.; Sato, Y.; Sata, M.; Kage, M.; Sugie, H.; Nunoi, K. Intermittent and Recurrent Hepatomegaly Due to Glycogen Storage in a Patient with Type 1 Diabetes: Genetic Analysis of the Liver Glycogen Phosphorylase Gene (PYGL). Diabetes Res. Clin. Pract. 2004, 65, 175–182.
- Sherigar, J.M.; Darouichi, Y.; Guss, D.; Mohanty, S.R. An Unusual Presentation of Glycogenic Hepatopathy with Bridging Fibrosis. ACG Case Rep. J 2018, 5, e31.
- Harrison, S.A.; Brunt, E.M.; Goodman, Z.D.; Di Bisceglie, A.M. Diabetic Hepatosclerosis: Diabetic Microangiopathy of the Liver. Arch. Pathol. Lab. Med. 2006, 130, 27–32.
- Balakrishnan, M.; Garcia-Tsao, G.; Deng, Y.; Ciarleglio, M.; Jain, D. Hepatic Arteriolosclerosis: A Small-Vessel Complication of Diabetes and Hypertension. Am. J. Surg. Pathol. 2015, 39, 1000–1009.
- Lackner, C.; Gogg-Kamerer, M.; Zatloukal, K.; Stumptner, C.; Brunt, E.M.; Denk, H. Ballooned Hepatocytes in Steatohepatitis: The Value of Keratin Immunohistochemistry for Diagnosis. J. Hepatol. 2008, 48, 821–828.
- Gramlich, T.; Kleiner, D.E.; McCullough, A.J.; Matteoni, C.A.; Boparai, N.; Younossi, Z.M. Pathologic Features Associated with Fibrosis in Nonalcoholic Fatty Liver Disease. Hum. Pathol. 2004, 35, 196–199.
- Nozaki, Y.; Petersen, M.C.; Zhang, D.; Vatner, D.F.; Perry, R.J.; Abulizi, A.; Haedersdal, S.; Zhang, X.-M.; Butrico, G.M.; Samuel, V.T.; et al. Metabolic Control Analysis of Hepatic Glycogen Synthesis In Vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 8166–8176.
- Krssak, M.; Brehm, A.; Bernroider, E.; Anderwald, C.; Nowotny, P.; Dalla Man, C.; Cobelli, C.; Cline, G.W.; Shulman, G.I.; Waldhäusl, W.; et al. Alterations in Postprandial Hepatic Glycogen Metabolism in Type 2 Diabetes. Diabetes 2004, 53, 3048–3056.
- Dongiovanni, P.; Meroni, M.; Mancina, R.M.; Baselli, G.; Rametta, R.; Pelusi, S.; Männistö, V.; Fracanzani, A.L.; Badiali, S.; Miele, L.; et al. Protein Phosphatase 1 Regulatory Subunit 3B Gene Variation Protects against Hepatic Fat Accumulation and Fibrosis in Individuals at High Risk of Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2018, 2, 666–675.
- Stender, S.; Smagris, E.; Lauridsen, B.K.; Kofoed, K.F.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Pennacchio, L.A.; Dickel, D.E.; Cohen, J.C.; Hobbs, H.H. Relationship between Genetic Variation at PPP1R3B and Levels of Liver Glycogen and Triglyceride. Hepatology 2018, 67, 2182–2195.
- Ozen, H. Glycogen Storage Diseases: New Perspectives. World J. Gastroenterol. 2007, 13, 2541–2553.
- Wright, T.L.F.; Umaña, L.A.; Ramirez, C.M. Update on Glycogen Storage Disease: Primary Hepatic Involvement. Curr. Opin. Pediatr. 2022, 34, 496–502.
- Massese, M.; Tagliaferri, F.; Dionisi-Vici, C.; Maiorana, A. Glycogen Storage Diseases with Liver Involvement: A Literature Review of GSD Type 0, IV, VI, IX and XI. Orphanet. J. Rare Dis. 2022, 17, 241.
- Torbenson, M.S. Genetic Diseases of the Liver. In Biopsy Interpretation of the Liver, 4th ed.; Wolters Kluwer: Philadelphia, PA, USA, 2022; pp. 546–552.
- Derks, T.G.J.; van Rijn, M. Lipids in Hepatic Glycogen Storage Diseases: Pathophysiology, Monitoring of Dietary Management and Future Directions. J. Inherit. Metab. Dis. 2015, 38, 537–543.