Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Iga Schachta and Version 2 by Catherine Yang.
Factor VII activating protease (FSAP) is produced mainly by the liver, which was shown both in animal and human studies. Besides hepatic sources, FSAP messenger RNA (mRNA) has been found in the murine [7] and human kidney, human pancreas and skeletal muscle. FSAP can undergo autoactivation, but various factors in vitro and in vivo can accelerate or inhibit the FSAP autoactivation process.
- factor VII activating protease
- coagulation
- hemostasis
1. Enhancers of scFSAP Activation
Over the years, several studies have shown that scFSAP activation is accelerated by heparin, a negatively charged glycosaminoglycan (GAG) [1][2][3][4][5][6][7][8][9][10][11][12][9,10,11,17,18,38,39,48,51,52,53,64]. The pioneering study by Etscheid et al. was the first to describe the enhancing effect of unfractionated heparin (UH) on the isolated FSAP autoactivation [3][11]. Various groups of researchers have performed mostly in vitro studies using purified or commercially available molecules [1][2][3][4][5][6][8][9][10][11][12][9,10,11,17,18,38,48,51,52,53,64]. It was demonstrated that UH [8][10][48,52], mast cell heparin, and low molecular weight heparin (LMWH) could bind to isolated FSAP [8][48]. These molecules propagate the autoactivation of isolated FSAP to varying degrees [8][48]. In contrast to UH [8][48] or native heparin [1][8][9,48], low molecular weight heparin (LMWH) had a minimal ability to act as the enhancer of isolated FSAP autoactivation. Moreover, LMWH specifically reversed the effect of spermidine, another propagator of tcFSAP generation, by inhibiting the spermidine-induced in vitro autoactivation of FSAP [1][9]. In turn, mast cell-derived heparin, which has a higher negative charge than UH, was more potent in terms of FSAP autoactivation increase in relation to UH [8][48]. The final conclusion of Muhl et al. was that the size, charge density, and conformational flexibility of polyanions appeared to be important for the interaction with isolated FSAP [8][48].
Contrary to the expectations based on the studies with purified systems, heparin failed to improve FSAP activation in human plasma [2][13][14][10,59,65]. Taking into consideration that sulfated glycosaminoglycans construct the extracellular matrix and are expressed by the endothelium [15][66], the ultimate role of heparin in FSAP autoactivation, especially in vivo, remains vague.
Importantly, FSAP activation could be influenced by the fibrinolysis regulator, uPA [6][38]. The generation of tcFSAP from purified scFSAP was accelerated by three forms of uPA, including uPA zymogen (single-chain uPA, scuPA), active uPA (two-chain uPA, tcuPA), and high molecular mass uPA (HMMuPA) [6][38]. Other studied isoforms, namely low molecular weight uPA, noncleavable and enzymatically inactive mutant scuPA-Gly158, and N-terminal fragment of high-molecular-mass urokinase (ATF) did not affect the activation of FSAP [6][38]. It indicated that the intact molecule and function of uPA were key to interacting with FSAP [6][38].
Choi-Miura et al. observed tcFSAP in the plasma of mice after hepatic failure and after partial hepatectomy [16][7]. To theour knowledge, this study was the first to indicate a connection between FSAP activation and tissue injury [16][7]. The incubation of human plasma [17][18][43,67] or human serum [19][68] with apoptotic [17][18][43,67] or necrotic cells [17][19][20][40,43,68] caused the binding of dead cells to FSAP [17][43] followed by the activation of FSAP [17][18][19][20][40,43,67,68]. No significant activation of plasma FSAP by living human T lymphocyte cells (Jurkat cells) was seen [17][43]. However, the possibility of successful FSAP activation may depend on the cell culture, because Kannemeier et al. described that isolated scFSAP autoactivated both in the absence and presence of murine VSMC [21][28].
Some studies have focused on specific molecules that could emerge from dead cells and contribute to the generation of tcFSAP. In the purified system, highly cationic histones [2][22][10,63] and their subtypes promoted scFSAP autoactivation [2][10]. The strongest effect was demonstrated by histone 3 (H3), H2A, and H4 [2][10]. For example, the internal sequence of amino acid residues of H2A has been demonstrated to be cardinal for the acceleration of FSAP autoactivation [2][10]. Histones proved to be the powerful propagators of the activation of endogenous FSAP as was shown in human plasma [2][13][15][23][24][10,59,66,69,70] and sera [22][63] samples of healthy individuals. Animal studies support the activation of FSAP by histones [2][10]. The activation of FSAP in mice via the injection of histones [2][10] appears to be a breakthrough. This finding provided evidence of histone-promoted FSAP autoactivation occurring in vivo [2][10]. Based on both animal and human studies on sepsis, it was hypothesized the in vivo activation of FSAP could be due to the freed histones, but at that point, no unequivocal proof of this conception was demonstrated [2][22][10,63]. To confirm this hypothesis, further experiments and human studies are needed.
It is worth remembering that, besides free histone fractions, they can be complexed with DNA in various forms, namely, nucleosomes, chromatin, or neutrophil extracellular traps (NETs) [15][22][25][63,66,71]. It is not clear in which form histones are released from the damaged cell [22][63]. This can be critical in the context of tcFSAP formation, as not all histone forms lead to FSAP activation with comparable efficiency [13][15][22][25][59,63,66,71]. According to Semeraro et al., the DNA-histone complex promoted less effective plasma FSAP activation in vitro relative to the histones alone [15][66]. Marsman et al. focused on the FSAP activation by nucleosomes [22][63], which consist of DNA wrapped around a histone octamer. If the nucleosomes were predigested to release histones and then added to human serum, tcFSAP was detected [22][63]. However, after the incubation of the intact nucleosomes with the serum, the activation of FSAP was much weaker [22][63]. Presumably, the negatively charged DNA neutralized the positive charge of histones [22][63], impeding them to promote FSAP autoactivation.
Notwithstanding, the formation of tcFSAP in plasma correlated with the nucleosome levels in adults post-surgery (r = 0.55, p < 0.0001 and r = 0.64, p < 0.0001 depending on the measurement method of FSAP activation); patients with severe sepsis and septic shock (r = 0.43, p = 0.006 and r = 0.44, p = 0.004, depending on the measurement method of FSAP activation); children with meningococcal sepsis (r = 0.72, p < 0.0001 and r = 0.62, p < 0.0001 depending on the measurement method of FSAP activation) [17][43]; and polytrauma patients (r = 0.76, p < 0.001) [13][59]. FSAP activation in plasma correlated also with the nucleosome release in melioidosis subjects (r = 0.74, p < 0.0001) [26][61]. These reports [13][17][26][43,59,61] suggest a link between FSAP and nucleosomes, but they do not clarify the mechanism by which nucleosomes would activate FSAP.
Interestingly, the treatment of human plasma with chromatin, a more complex organization of nucleosomes, led to the activation of endogenous FSAP in vitro [13][59]. The histones, together with the chromatin DNA, are also components of NETs [25][71]. The formation of NETs is a type of cell death called NETosis, and it involves the release of nuclear factors [25][71]. Analogically to the nucleosomes [22][63], when purified FSAP or plasma was added to the neutrophils with induced NETosis, the binding between FSAP and NETs was seen; however, NETs formed during NETosis failed to alter FSAP activation in vitro [25][71]. Only if NETs were degraded by DNase and histones were freed, scFSAP conversion to tcFSAP in human plasma became significantly promoted [25][71]. As in nucleosomes [22][63], in NETs, DNA seemed to neutralize the histone functionality in the context of FSAP autoactivation [25][71]. Thus, in the case of more complex structures consisting of histones, histone accessibility seems to be essential to promote FSAP autoactivation.
Spermidine and spermine, which are positively charged polyamine compounds, can be released massively to the plasma during tissue injury or cellular death [1][9]. Spermidine increased intermolecular association between scFSAP and scFSAP, scFSAP and tcFSAP, as well as tcFSAP and tcFSAP [1][9]. Both spermidine and spermine enhanced FSAP autoactivation in vitro [1][9]. This feature is not equally presented by other tested polyamines, including putrescine, which exhibited little stimulatory function towards FSAP autoactivation [1][9]. Polyamines are elevated in the inflammatory tissues and malignant cells [1][9]. Thus, a rupture of such cells could possibly provide enough polyamines to facilitate FSAP autoactivation in vivo.
Nucleic acids, such as RNA, can also be released to the plasma as a result of cell damage [27][26]. Nakazawa et al. identified negatively charged extracellular RNA as the cell-derived cofactor for scFSAP autoactivation in vitro [27][26]. RNA bound both to scFSAP at multiple sites of the heavy and light chain and to tcFSAP within its heavy chain [27][26]. Important domains of FSAP enabling interaction with RNA are EGF2 and EGF3 [9][51]. FSAP at levels close to physiological values created RNA-FSAP complexes in vitro [27][26]; however, the minimal necessary fragment length of RNA to serve as a cofactor is 100 [27][26]–200 nucleotides [9][51]. FSAP autoactivation was accelerated by ribosomal RNA (rRNA) [9][27][26,51], mRNA [9][51], transfer RNA (tRNA), bacterial and viral RNA, as well as artificial RNA [27][26] in the purified system [9][27][26,51]. Notwithstanding, the study of Altincicek et al. showed that tRNA could augment the conversion of scFSAP to tcFSAP in vitro, but only under the condition of high levels of tRNA [9][51]. The first doubts about the significance of RNA in FSAP activation originated from the experiments reported by Zeerleder et al. [20][40] and Stephan et al. [17][43]. These studies showed the contribution of apoptotic and necrotic cells to generate tcFSAP in vitro [17][20][40,43]. The authors determined which cell-derived structure could be responsible for their observations [17][20][40,43]. RNA was excluded as an accelerator of FSAP activation because there was no difference in results between RNase-treated and untreated cells [17][20][40,43]. Two independent studies by Yamamichi et al. confirmed that the incubation of isolated scFSAP with RNA promoted FSAP autoactivation in the purified system [1][2][9,10], but there was no such effect existing in the human plasma [2][10].
Finally, there are contrasting data on DNA and FSAP activation. Semeraro et al. showed that DNA alone was not able to generate tcFSAP in the plasma [15][66]. Neither DNA homologue [27][26] nor DNA [25][71] was able to affect tcFSAP generation. DNA also did not form a complex with FSAP [27][26]. Conversely, Altincicek et al. noted that DNA had to be added at high levels to promote FSAP activation [9][51]. Apparently, the capability of nucleic acids to enhance FSAP autoactivation is controversial.
As FSAP autoactivation could be promoted by charged molecules, Sperling et al. conducted a study of endogenous FSAP activation on the extended planar material surfaces [28][50]. The authors demonstrated that cationic polyethylenimine (PEI) could induce FSAP autoactivation in the plasma and in whole blood samples [28][50]. Plasma FSAP activation was also seen on positively charged poly-L-lysine (PLL), but the effect was weaker in comparison to PEI [28][50]. FSAP activation was not present on the surface with negatively charged substrates such as glass and self-assembled monolayer with a carboxyl group (C(=O)OH), or with a neutral charge such as polytetrafluoroethylene (PTFE, teflon) [28][50]. Sperling et al. discussed that the higher activation of FSAP on PEI than on PLL can indicate how significant the charge density and chemical structure are for FSAP autoactivation [28][50]. It was concluded that cationic surfaces have comparable functionality to cationic macromolecules such as histones in terms of FSAP autoactivation [28][50]. As mentioned, there are negatively charged molecules that promote FSAP autoactivation in the purified system [1][2][3][4][5][6][8][9][10][11][12][27][9,10,11,17,18,26,38,48,51,52,53,64] though neither anionic molecules [2][13][14][10,59,65] nor extended flat anionic surfaces induced FSAP autoactivation in plasma [28][50]. The possible reasons for these observations were considered, namely an inadequate molecular conformation, the charge density of the anionic surface, or interfering plasma proteins [28][50]. Overall, this study is especially intriguing in terms of the safety of polycationic surfaces in medical devices and implants, as they could possibly lead to the generation of tcFSAP [28][50].
As presented, many various molecules are known as propagators of FSAP activation. Interestingly, two theoretical mechanisms of FSAP autoactivation have been proposed so far: the scaffold (template) mechanism [1][9][27][28][9,26,50,51] and the autoactivation complex model [1][2][28][9,10,50]. They are presented in Figure 12. The type of model depends on the positive or negative charge of the enhancer of FSAP autoactivation [1][28][9,50]. Both autoactivation mechanisms engage the NTR and EGF3 domains of FSAP [1][28][9,50]. Before the exploration of the details of these two models, it is important to understand the interactions between the domains of scFSAP. It was summarized that within the single inactive scFSAP molecule, the basic amino acids of the EGF3 domain (Arg144, His145, Lys146, Arg147, Arg148, Ser149, and Lys150) are guarded by the acidic (Glu8, Asp11, Asp13, Asp17, Asp20, Glu24, Asp25, Glu29, Glu30, Glu40, Asp43, Glu48, Asp49, and Asp52) and aromatic residues (Trp14, Tyr19, Tyr21, Tyr23, Tyr26, Trp44, Tyr45, and Tyr46) in the NTR [1][9]. This intramolecular interaction between positively charged EGF3 and negatively charged NTR protects scFSAP from binding to another scFSAP and thus prevents FSAP autoactivation [1][28][9,50]. However, the presence of some ionized molecules [1][28][9,50] or surfaces [28][50] can unblock EGF3 from NTR, making FSAP vulnerable to intermolecular autoactivation.
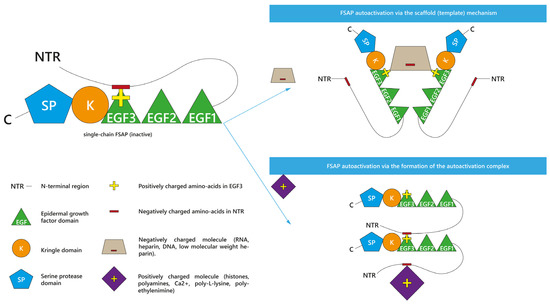
Figure 12. Two theoretical models of factor VII activating protease (FSAP) autoactivation. Negatively charged molecules promote FSAP autoactivation in vitro in the scaffold (template) mechanism or the interaction by binding. Positively charged molecules form the autoactivation complex together with FSAP molecules.
Irrespective of the results in plasma [2][13][14][10,59,65] and the study on anionic surfaces [28][50], negatively charged RNA [1][9][27][9,26,51] and heparin [1][9][9,51] promoted FSAP autoactivation in the purified system in the so-called scaffold (template) mechanism [1][9][27][9,26,51] or the interaction by binding [28][50]. In this model, an anionic molecule binds with the positively charged [28][50] EGF3 domains of different scFSAP molecules, forming a specific anionic bridge [1][27][28][9,26,50] between them. This anionic scaffold linking scFSAP molecules would facilitate their autoactivation [1][28][9,50]. As mentioned, LMWH had limited capability to accelerate FSAP autoactivation [1][8][9,48]. The possible reason could be surface of LMWH is too small to create a scaffold between two FSAP molecules [1][9].
The autoactivation complex is the second proposed mechanism [1][2][28][9,10,50]. It is based on the interaction with the cationic molecules, such as histones [2][10], and polyamines such as spermidine, spermine, or putrescine [1][9]. Because spermidine [1][9] and H3 [2][10] elevated the intermolecular binding of scFSAP, it was proposed that the positively charged molecules could form an autoactivation complex together with FSAP molecules [1][9]. More precisely, the presence of cationic molecules near scFSAP would lead to the release of the negatively charged NTR from EGF3 domain [1][28][9,50]. Thus, cationic molecules would allow for the intermolecular interaction of the released NTR of one FSAP molecule with the EGF3 of another FSAP molecule, leading to FSAP autoactivation [1][28][9,50]. This mechanism shows cationic molecules as promoters of the FSAP autoactivation complex [1][28][9,50].
It shall be highlighted that both the autoactivation complex and template mechanism are theoretical models and require further research.
2. Inhibitors of scFSAP and Its Activation
Contrary to the propagation of scFSAP autoactivation, little is known about its inhibition. It remains obscure whether any natural inhibitor present in human blood is able to interact with and inhibit scFSAP as the data on this subject is fragmentary and, in some cases, contradictory. Wygrecka et al. even suggested that an effective interaction of the protease inhibitors with scFSAP is not expected due to the proteolytical inactivity of this form [7][39].
Serpins circulating in plasma at high levels are C1-inhibitor (C1-inh) and α-2-antiplasmin (AP) [2][10], but it is not known if they inhibit scFSAP activation. Shortly after FSAP discovery, Etscheid et al. indicated that AP and C1-inh caused a slower autoactivation of purified scFSAP [3][11]; however, to the best of theour knowledge, it is the only study to provide such information. Furthermore, even the authors of this manuscript suspected that AP- and C1-inh-driven slowdown of FSAP activation was affected by the rapid inactivation of tcFSAP generated during the assay [3][11] instead of scFSAP. This assumption is in agreement with the common interpretation of the detected FSAP-inhibitor binding. Namely, the complexes between FSAP and AP [2][13][17][18][19][22][23][24][25][26][28][29][10,43,50,59,61,62,63,67,68,69,70,71] or C1-inh [17][18][24][43,67,70] are considered the markers of FSAP autoactivation in various in vitro and in vivo studies. Thus, although AP and C1-inh are not usually associated with scFSAP inhibition, their complexing with FSAP refers to the level of formed tcFSAP that is bound with the protease inhibitors.
An ambiguous observation concerning C1-inh, AP, and scFSAP can be found in the study by Kanse et al. [13][59]. In human plasma of healthy individuals, FSAP was co-immunoprecipitated not only with AP and C1-inh but also with many other inhibitors such as α2-macroglobulin, α1-trypsin inhibitor, and heparin cofactor 2 [13][59]. The significance of that result was not further examined, although the authors could not exclude that the mentioned inhibitors made complexes in vivo with scFSAP as the main FSAP form circulating in the plasma [13][59]. To verify this hypothesis, further studies should be conducted.
In the case of PAI-1, another member of the serpin family, it is also not clear whether it can inhibit the formation of tcFSAP from scFSAP [7][39]. Wygrecka et al. reported that PAI-1 did not bind or only exhibited limited binding with isolated scFSAP [7][39]. Moreover, this small level of FSAP-PAI-1 complexes could in fact be the result of the interaction between PAI-1 and tcFSAP, which autoactivated from scFSAP during the experiment [7][39].
As mentioned previously, LMWH is a weak enhancer of FSAP autoactivation [8][48], but it inhibits spermidine-induced FSAP autoactivation [1][9]. Table 14 summarizes FSAP activation inhibitors described in this subsection.
Table 14.
Single-chain factor VII activating protease (scFSAP) activation inhibitors for which data are contradictory.
| Name | Studies In Vitro Describing the Influence | Contradictory Data |
|---|---|---|
| α-2-antiplasmin (AP) | [3][11] | FSAP-AP complexes are considered as the marker of completed FSAP activation [2][13][17][,4318,50],59[19][22][23][24],61[,6225,63][,6726,68],69[28][29][10,70,71]. |
| C1-esteraze inhibitor (C1-inh) | [3][11] | FSAP-C1-inh complexes are considered as the marker of completed FSAP activation [17][18][24][43,67,70]. |
| Plasminogen activator inhibitor type 1 (PAI-1) | Limited scFSAP-PAI-1 binding [7][39]. | Another experiment of the same authors indicated that scFSAP did not bind with PAI-1. |
| Low molecular weight heparin (LMWH) | LMWH inhibited spermidine-induced FSAP autoactivation [1][9]. | LMWH weakly promotes FSAP activation [8][48]. |