The liver is a major store of glycogen and is essential in maintaining systemic glucose homeostasis. In healthy individuals, glycogen synthesis and breakdown in the liver are tightly regulated. Abnormal glycogen metabolism results in prominent pathological changes in the liver, often manifesting as hepatic glycogenosis or glycogen inclusions. This can occur in genetic glycogen storage disease or acquired conditions with insulin dysregulation such as diabetes mellitus and non-alcoholic fatty liver disease or medication effects.
Excessive glycogen accumulation within hepatocytes occurs in diseases caused or accompanied by the dysregulation of carbohydrate metabolism, as well as in conditions not primarily driven by altered carbohydrate metabolism. In these pathological conditions, the excess glycogen is distinctly visible on H&E; the hepatocytes usually exhibit cytoplasmic pallor and rarefaction or may show cytoplasmic-glycogen-filled inclusion bodies. Some hepatocytes may also demonstrate glycogen-filled nuclear vacuoles without a delimiting membrane (“glycogenated nuclei”); these are frequently seen in patients with diabetes and obesity, although they can also be seen in other liver conditions such as Wilson’s disease.
1. Glycogenic Hepatopathy
Glycogenic hepatopathy is a distinctive, reversible clinicopathological entity caused by excessive overloading of hepatocytes with glycogen, classically seen in patients with poorly controlled type 1 diabetes mellitus. While first documented as a component of Mauriac syndrome in 1930
[1], it was subsequently recognized that glycogen overloading of the liver can occur without the associated findings of growth failure, delayed puberty, and cushingoid features. Previously referred to variably as hepatic/liver glycogenosis
[2][3], liver glycogen storage
[4][5], or DM-associated glycogen storage hepatomegaly
[6], the term “glycogenic hepatopathy” was coined in 2006 in the first paper to systematically describe the histological findings and has now gained widespread acceptance and usage
[7].
1.1. Clinical Findings and Pathogenesis
Glycogenic hepatopathy typically occurs in children and adults with marked or prolonged hyperglycemia, followed by exogenous administration of insulin in the setting of type 1 DM. Most patients have hepatomegaly and elevated serum aminotransferases, which improve after improved glycemic control
[8]. Rarely, ascites may also be a presenting sign, but this too resolves with adequate control of blood sugar levels
[7][9]. In the current era of modern insulin regimens and intensive glycemic monitoring, it is rare for patients to present with the full complement of Mauriac syndrome
[7][10], although patients with glycogenic hepatopathy may have impaired growth compared to their peers
[11].
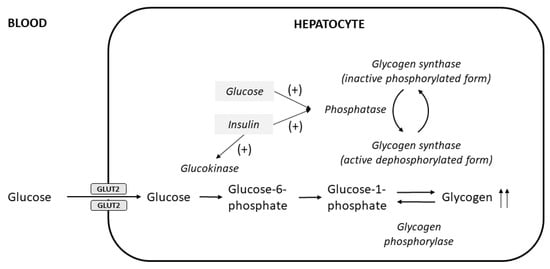
The excessive glycogen accumulates in hepatocytes because of the insulin-independent passage of glucose into hepatocytes during the period of hyperglycemia, followed by insulin-mediated conversion into hepatic glycogen; both elevated glucose and insulin are required
[12] (
Figure 12). Patients who take excess insulin and are subsequently treated with glucose to correct hypoglycemia can also develop glycogenic hepatopathy
[13]. Glycogenic hepatopathy has only rarely been reported in patients with type 2 DM
[14]; the insulin resistance underlying type 2 DM typically leads to reduced hepatic glycogen synthesis and increased lipogenesis, manifesting predominantly as non-alcoholic fatty liver disease
[15]. Nevertheless, glycogenic hepatocytes can also be present in these patients
[16].
Figure 12. Pathogenesis of hepatocellular glycogen accumulation in the liver, highlighting the role of both elevated glucose levels and insulin treatment in glycogenic hepatopathy (modified from Munns et al.
[12]). Glucose enters hepatocytes via facilitated diffusion through glucose transporter 2 (GLUT2) independent of insulin, where it is then phosphorylated to glucose-6-phosphate by glucokinase and subsequently converted to glycogen by glycogen synthase. Insulin activates glucokinase, and also acts together with elevated glucose levels to stimulate the phosphatase enzyme to produce the active dephosphorylated form of glycogen synthase
[17].
Apart from type 1 DM, glycogenic hepatopathy has also been described in other clinical conditions, such as following the administration of short-term high-dose steroid therapy
[18], dumping syndrome
[19], and anorexia nervosa
[20][21]. The former two conditions result in a hyperglycemic–hyperinsulinemic state similar to that observed in type 1 DM patients. The mechanism behind excessive hepatic glycogen accumulation in anorexia nervosa is uncertain; it may be related to the body’s adaptation to starvation by sparing glucose utilization and stimulation of hepatic glycogenesis in order to prevent severe hypoglycemia and death
[20][21].
A case–control study confirmed that patients with glycogenic hepatopathy had poorer glycemic control, as evidenced by a history of recurrent episodes of diabetic ketoacidosis and elevated HbA
1c levels
[10]. However, it is still uncertain why only a small subset of patients with type 1 DM develop glycogenic hepatopathy. The excess glycogen accumulated in the hepatocytes appears to have a normal molecular structure, and the activity of enzymes glucose-6-phosphatase, phosphorylase acid maltase, amylo-1,6-glucosidase, and phosphoglucomutase appears largely preserved
[22]. Although a heterozygous mutation in liver glycogen phosphorylase kinase was found in one patient with type 1 DM and Mauriac syndrome
[23], this does not account for the vast majority of cases in which glycogenic hepatopathy is a reversible phenomenon. It may be that single nucleotide polymorphisms in genes involved in glycogen metabolism predispose certain individuals to developing or manifesting the disease
[24].
1.2. Pathological Findings
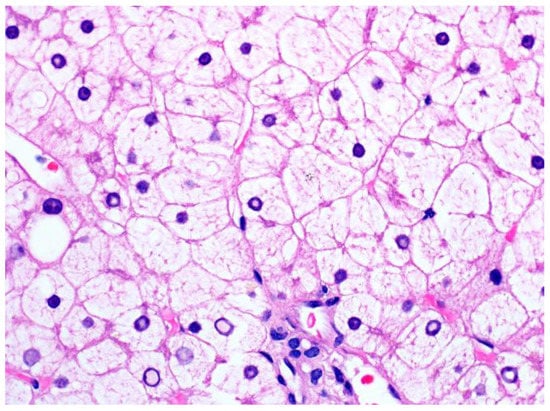
Histologically, glycogenic hepatopathy is characterized by a striking diffuse hepatocellular change—the hepatocytes appear pale with cytoplasmic rarefaction and accentuated cell membranes, due to cytoplasmic accumulation of glycogen (Figure 23). PAS and PAS-D stains can be used to prove that the clearing of the cytoplasm is due to glycogen, but needs to be interpreted in conjunction with H&E and clinical findings, given that normal hepatocytes have abundant baseline amounts of glycogen. Glycogenated nuclei can also be present but are a nonspecific finding. The swollen hepatocytes compress the sinusoids, imparting a “paved” appearance. No disruption of the lobular architecture and no or only minimal inflammation is seen. Macrovesicular steatosis, usually mild to moderate, may also be present in some cases.
Figure 23. A liver biopsy from a patient with glycogenic hepatopathy demonstrating swollen hepatocytes with abundant clear cytoplasm due to glycogen accumulation (H&E stain; original magnification 600×). Occasional glycogenated nuclei can be seen.
Fibrosis is usually not a feature of glycogenic hepatopathy. Although occasional cases may have mild portal or pericellular fibrosis
[7], only very rare cases of bridging fibrosis have been reported
[11][25]. If there is significant fibrosis, other conditions such as steatohepatitis or diabetic hepatosclerosis should be excluded. Diabetic hepatosclerosis occurs in some patients with long-standing diabetes mellitus; it is characterized by extensive, dense perisinusoidal fibrosis in the absence of steatohepatitis
[26]. Diabetic hepatosclerosis appears to be a microangiopathic disease of the liver and is often accompanied by hyaline arteriolosclerosis in the liver
[27].
2. Glycogenosis in NAFLD
Recently, it has been recognized that excess accumulation of glycogen within hepatocytes can also be seen in the context of non-alcoholic fatty liver disease. In these cases, the dominant clinicopathological picture is not that of the typical glycogenic hepatopathy patient (who has type 1 DM and poor glycemic control, hepatomegaly and elevated transaminases); instead, patients are typically older, have type 2 DM and other features of metabolic syndrome, and steatohepatitis. These glycogenic hepatocytes can be seen in up to 54% of NAFLD cases
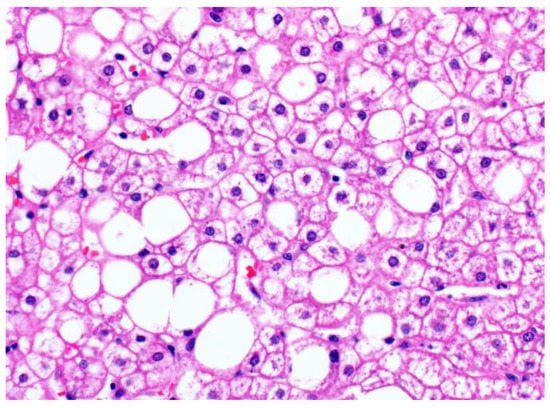
[16]; the glycogenated hepatocytes occur focally, often in patches in the centrilobular zone (
Figure 34). Even when glycogenated hepatocytes are present more extensively, the liver tissue in patients with NAFLD does not show the same diffuse striking hepatocellular changes characteristic of glycogenic hepatopathy. Individually, the enlarged, pale, swollen hepatocytes appear similar to those seen in glycogenic hepatopathy but lack the diffuse distribution of glycogenic hepatopathy. They may also be confused with ballooned hepatocytes, a form of hepatocyte injury with cytoskeletal alterations
[28] that is used to distinguish steatohepatitis from simple steatosis
[29]. Glycogenated nuclei may also be seen.
Figure 34. Patchy glycogenosis in NAFLD (original magnification 400×). The empty rounded spaces are large lipid droplets displacing the cell nuclei, while the glycogenated hepatocytes show cytoplasmic pallor but retain the central position of the nuclei and angular cell contours.
The presence of focal/patchy hepatocyte glycogenosis in a significant proportion of NAFLD patients underscores the complexity of glucose regulation. Insulin resistance, which is a major hallmark of NAFLD, impairs hepatic glycogen synthesis through dysregulation of glucokinase translocation from the nucleus to the cytoplasm, a rate-controlling step in insulin-stimulated hepatic glycogen synthesis
[30]. Hepatocytes in patients with type 2 DM, characterized by insulin resistance, similarly have decreased glycogen content
[31]. It is likely that dysregulation of the closely interrelated carbohydrate and lipid metabolic pathways in the liver can cause glycogenosis in this setting when insulin-mediated pathways are bypassed, leading to shunting of substrates from one path to another. Individuals at high risk of NAFLD who have a genetic variation in
PPP1R3B, a hepatic glycogen metabolism regulatory protein involved in glycogen synthesis, have been reported to have reduced hepatic steatosis
[32], possibly due to shunting of glucose to glycogen synthesis and reduction of de novo lipogenesis
[33]. Indeed, glycogenosis in the liver biopsies of NAFLD patients appears to be associated with decreased steatosis
[16].
3. Glycogen Storage Disease
Glycogen storage diseases (GSDs) are genetic disorders of glycogen metabolism, many of which present as an abnormal accumulation of glycogen or lipid in the liver. They are classified based on enzyme deficiency and the affected tissue (muscle and liver); the GSDs primarily affecting the liver are types 0, I, III, IV, VI, and IX
[34]. The hepatic GSDs are generally characterized by hepatomegaly and fasting hypoglycemia, although the individual clinical and biochemical profiles will differ depending on the specific enzymatic defect
[35] (
Table 1). Readers may refer to recent comprehensive reviews on this topic for more details
[35][36].
Table 1.
Glycogen storage disorders that primarily affect the liver
.
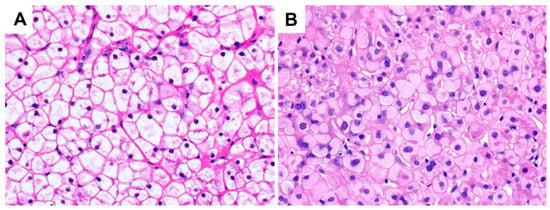
The glycogenosis seen on liver biopsies in patients with hepatic GSD can look indistinguishable from that seen in glycogenic hepatopathy, although in some cases of GSD, there may be subtle but increased cytoplasmic clumping of glycogen, and in type IV there are distinctive ground-glass type inclusions (Figure 45). The clinical setting (the absence of poorly-controlled diabetes) is key to arriving at the correct diagnosis, with subsequent confirmation by additional biochemical assays or sequencing.
Figure 45. Glycogen storage disease (original magnification 400×): (A) type IX, with a similar histological appearance to glycogenic hepatopathy; (B) type IV, demonstrating hepatocellular ground-glass type cytoplasmic inclusions. These PAS-positive inclusions may be diastase-resistant as they comprise amylopectin-like material rather than typical glycogen. Chronic hepatitis B infection and drug-induced glycogen pseudoground-glass type changes can have a similar appearance and should be excluded.
Hepatic steatosis may also be present in patients with GSD. The dysregulation of liver glycogen metabolism often leads to compensatory increases in lipolysis from extrahepatic sources and mitochondrial fatty acid oxidation in GSD types associated with fasting ketotic hypoglycemia (0, III, VI, IX, and XI). In contrast, the hyperlipidemia seen in GSD type I results from the intracellular accumulation of glucose-6-phosphate, leading to increased glycolysis, increased production of acetyl-CoA, and increased lipogenesis
[38].