Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Lai Yue Chan.
Gomesin is a cationic antimicrobial peptide which is isolated from the haemocytes of the Brazilian tarantula Acanthoscurria gomesiana and can be produced chemically by Fmoc solid-phase peptide synthesis. Gomesin exhibits a range of biological activities, as demonstrated by its toxicity against therapeutically relevant pathogens such as Gram-positive or Gram-negative bacteria, fungi, cancer cells, and parasites.
- antimicrobial
- anticancer
- gomesin
- β-hairpin
- disulfide bonds
1. Introduction
Multidrug-resistant bacteria, also known as superbugs, have become a serious hazard to public health, and antimicrobial resistance has been declared to be one of the top ten global public health threats facing humanity. The social and economic impact of antimicrobial resistance is huge because of prolonged hospital stays and the need for expensive medications [1,2][1][2]. For example, there are more than 2.8 million antibiotic-resistant infections and more than 35,000 deaths each year in the United States [3]. As a result of resistance, traditional antibiotics are becoming increasingly ineffective, necessitating the development of better antimicrobial alternatives. In addition to existing treatment strategies, such as using conventional antibiotics used to target protein synthesis [4], antimicrobial peptides (AMPs) are thought to be a feasible alternative for eliminating multidrug-resistant bacteria because their mode of action differs from that of currently available antibiotics; they disturb cell membrane integrity [5,6][5][6], impede protein [7,8,9,10,11][7][8][9][10][11] and cell wall formation [12[12][13][14][15],13,14,15], and alter enzyme function [10,16,17][10][16][17]. AMPs can be used for simultaneous or subsequent treatments in many bacterial illnesses as they have independent therapeutic efficacies and fewer adverse effects than conventional antibiotics [18].
AMPs are found throughout nature and play a vital role in the innate immune systems of many species. They have a wide spectrum of biological activities (e.g., antimicrobial, antifungal, antiparasitic, and antiviral properties) and a wide range of secondary structures, as highlighted in Figure 1 [19]. With rising numbers of drug-resistant bacteria and an increasing awareness of the misuse of antibiotics, AMPs are being considered as alternative antimicrobial therapies and are a major focus of research across the world, with promising applications in health, food, animal husbandry, agriculture, and aquaculture. Mammals, amphibians, microbes, and insects are the main source of AMPs, according to statistics reported by Duwadi et al. [20]. AMPs discovered in aquatic animals (e.g., tachyplesin from Tachypleus tridentatus and magainin from Xenopus laevis) and terrestrial animals (e.g., androctonin from the scorpion Androctonus australis, and protegrins from porcine leukocytes) have also received much interest [21].
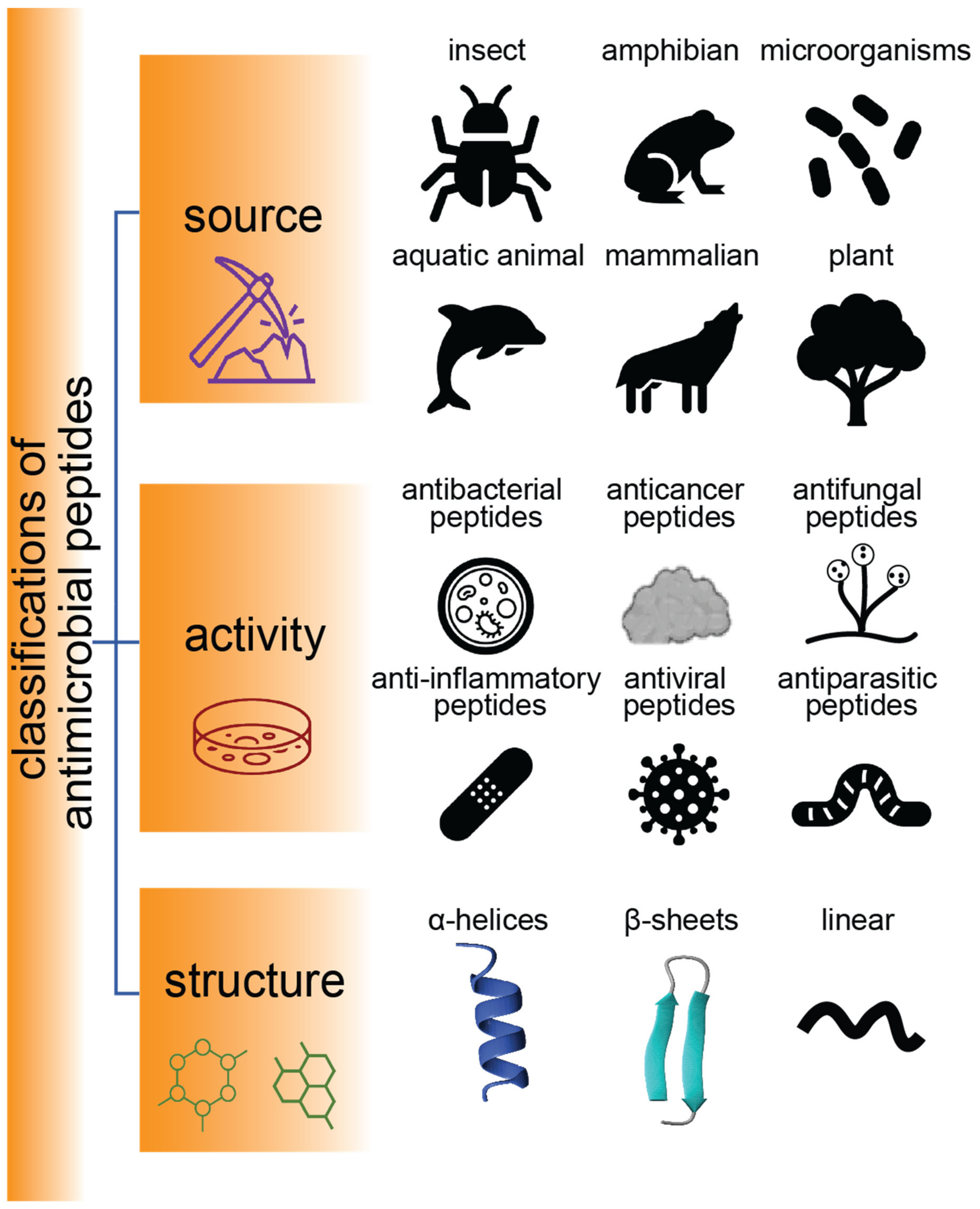
Figure 1.
Classification of AMPs. Schematic representation of sources, activities, and structures of AMPs.
Several classes of insect AMPs have been reviewed recently [22]. The current report focuses on a β-hairpin peptide known as gomesin (Gm), which is still relatively understudied. This 18-amino-acid antimicrobial peptide is isolated from the haemolymph of the tarantula spider, Acanthoscurria gomesiana, and inhibits bacterial growth as well as the development of filamentous fungus and yeast [23]. Apart from its antimicrobial activity, its cytotoxicity to cancer cells gives Gm anticancer properties. Given some of the drawbacks of current anticancer chemotherapy, including substantial side effects and/or the development of drug resistance, there is interest in exploring gomesin as an anticancer drug. To date, there are many anticancer drugs on the market, but only 10% are in the peptide category [24,25][24][25]. Since peptides are generally considered a safer alternative than traditional cytotoxic drugs, they have recently gained interest as potential anticancer therapeutics.
Since the discovery of Gm in 2000 [23], it has undergone more than 20 years of research, as summarised in Figure 2. To begin to understand its functions, the first three-dimensional solution structure of gomesin (PDB ID: 1KFP) was reported in 2002 [26], which was followed by a structure–activity relationship study in 2006 [27]. From 2007 to 2018, a range of bioactivities of Gm were characterised (including anticancer and antimalarial activities), and the chemical synthesis of a series of analogues with various chemical modifications were reported, including those involving amino acid mutations [28], with/without disulfide bonds [29], and backbone cyclization [30]. The first backbone cyclization study on gomesin was reported in 2013 and included a three-dimensional structure of the cyclic form [31]. Several structure–activity studies based on cyclic gomesin were reported thereafter, with a focus on optimizing antimicrobial and anticancer properties of cyclic gomesin analogues.
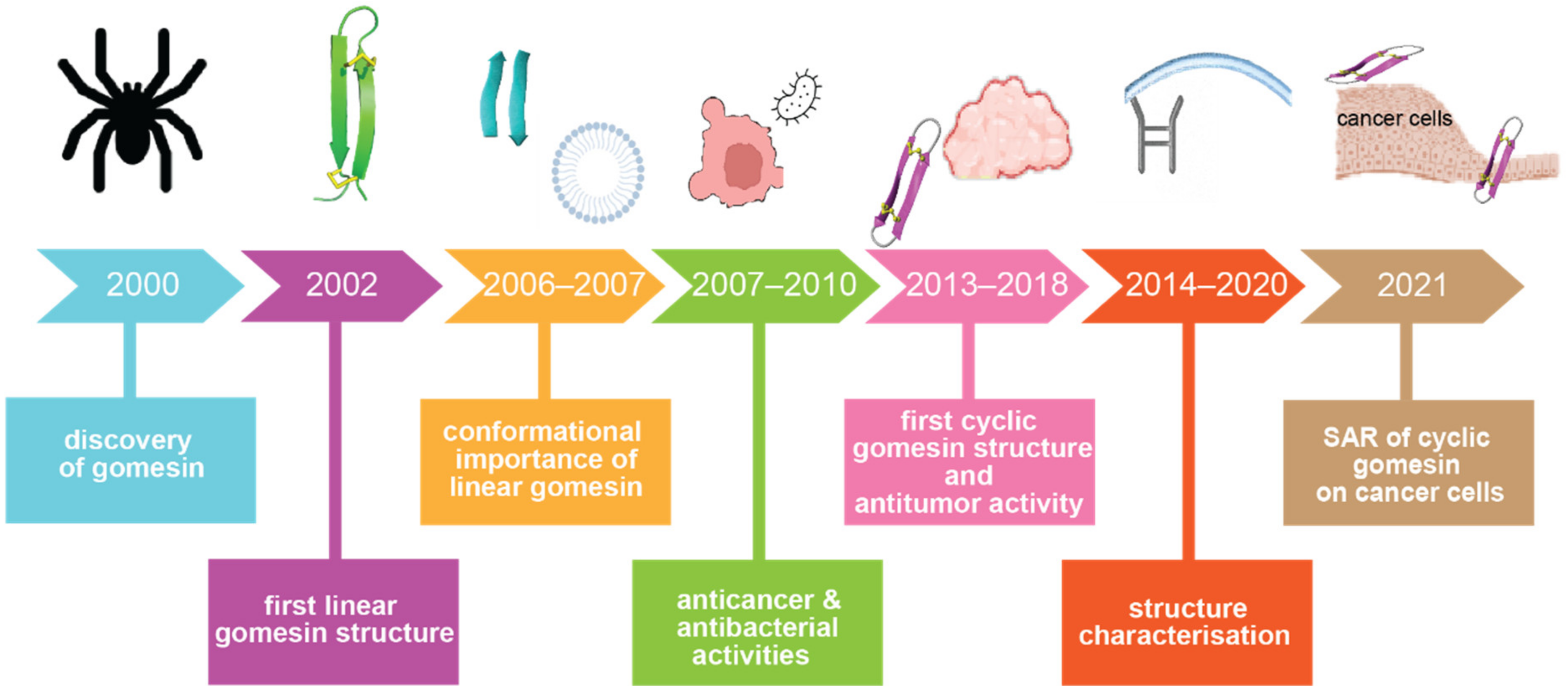
Figure 2. Historical timeline of gomesin studies. Key milestones in understanding the structure and activities of gomesin are indicated. Abbreviation: SAR: structure–activity relationship.
2. Discovery, Synthesis and Structural Characterization of Gomesin
Figure 3 schematically illustrates the steps involved in the isolation and structural characterization of gomesin. Through initial peptide sequencing and structural characterization, gomesin was reported to comprise 18 amino acids, including a pyroglutamic acid residue at the N-terminus, an amidated arginine at the C-terminus, and 4 cysteine residues that form 2 disulfide linkages (ZCRRLCYKQRCVTYCRGR-NH2), and with a molecular weight of 2270.4 Da [23]. In that first study Edman degradation was performed to determine the peptide sequence, but more recent studies of peptides typically use MS based peptide sequencing.
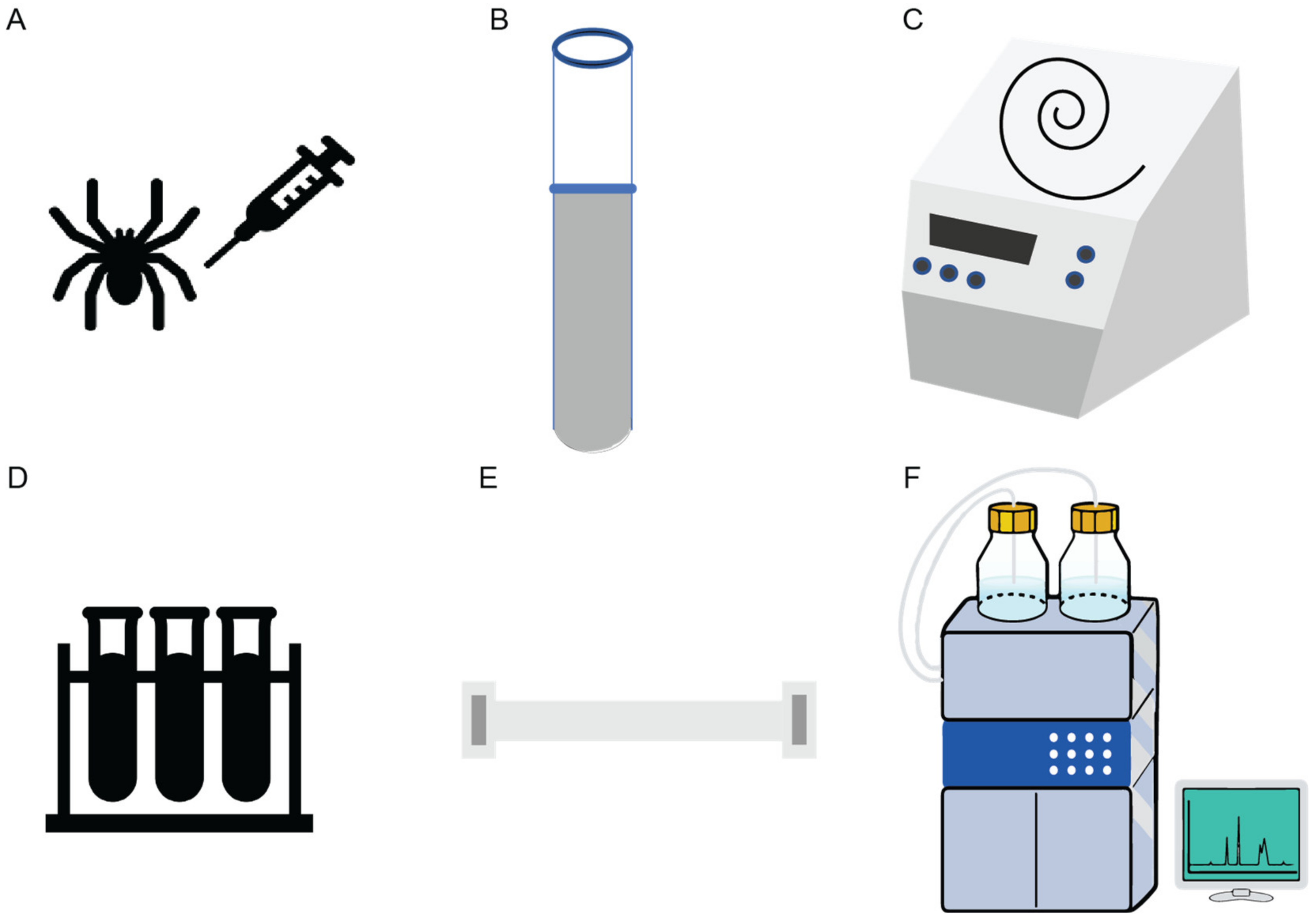
Figure 3. Schematic illustration of the isolation of Gm from spiders, hemolymph collection, and peptide extraction and purification. (A) Hemolymph collection from a spider is typically performed in sodium citrate buffer (pH 4.6). (B) Hemocytes are collected from the hemolymph. (C) Concentration and pyrolysis steps are done using vacuum centrifugation. (D) Solid phase extraction using pre-purified supernatant from ultrasonic treatment is then applied. (E) Aquapore RP-300 C8 column for peptide elution. (F) Further purification of the active peptide through high performance liquid chromatography-size exclusion column (HPLC-SEC1) completes the process [23].
While many early studies of gomesin involved the use of natively isolated peptide, the development of synthesis methods to make gomesin was important for establishing structure–activity relationships. In general, currently marketed therapeutic peptides, or those made for laboratory studies, are typically produced by either chemical synthesis or recombinant DNA technology [32]. So far solid-phase peptide synthesis (SPPS) is the most used method to produce Gm [27]. Using Fmoc (fluorenyl methoxycarbonyl) based protection chemistry, peptides are typically assembled from the carboxyl terminus (an amidated C-terminus in the case of gomesin) to the amino terminus (N-terminus with a pyro-glutamic acid residue in the case of gomesin) of the amino acid chain. By contrast, in a natural cell environment, peptides are synthesised from the N- to the C-terminus [33].
The amidated C-terminal arginine residue of gomesin has been reported to be important for minimizing enzymatic degradation, improving peptide stability, and with the potential to enhance antimicrobial activity due to the protonation of the sidechain of this C-terminal residue [20,34][20][34]. It is thus of interest to explore the prevalence of this charged C-terminal motif in other AMPs. Figure 4 shows a comparison of Gm with antimicrobial peptides from other arthropods (e.g., tachyplesin-Ⅰ [35[35][36],36], polyphemusin-II [37], androctonin [38]) and porcine leukocyte families (e.g., protegrin-Ⅰ [39]) and reveals high sequence and structural similarity. These peptides all comprise antiparallel β-sheets which contribute to their high stability profiles [40]. It is noteworthy that the N- and C-termini of these peptides are close to each other, leading to the possibility that they could be joined chemically to produce cyclic derivatives, as described later in this aentrticley.
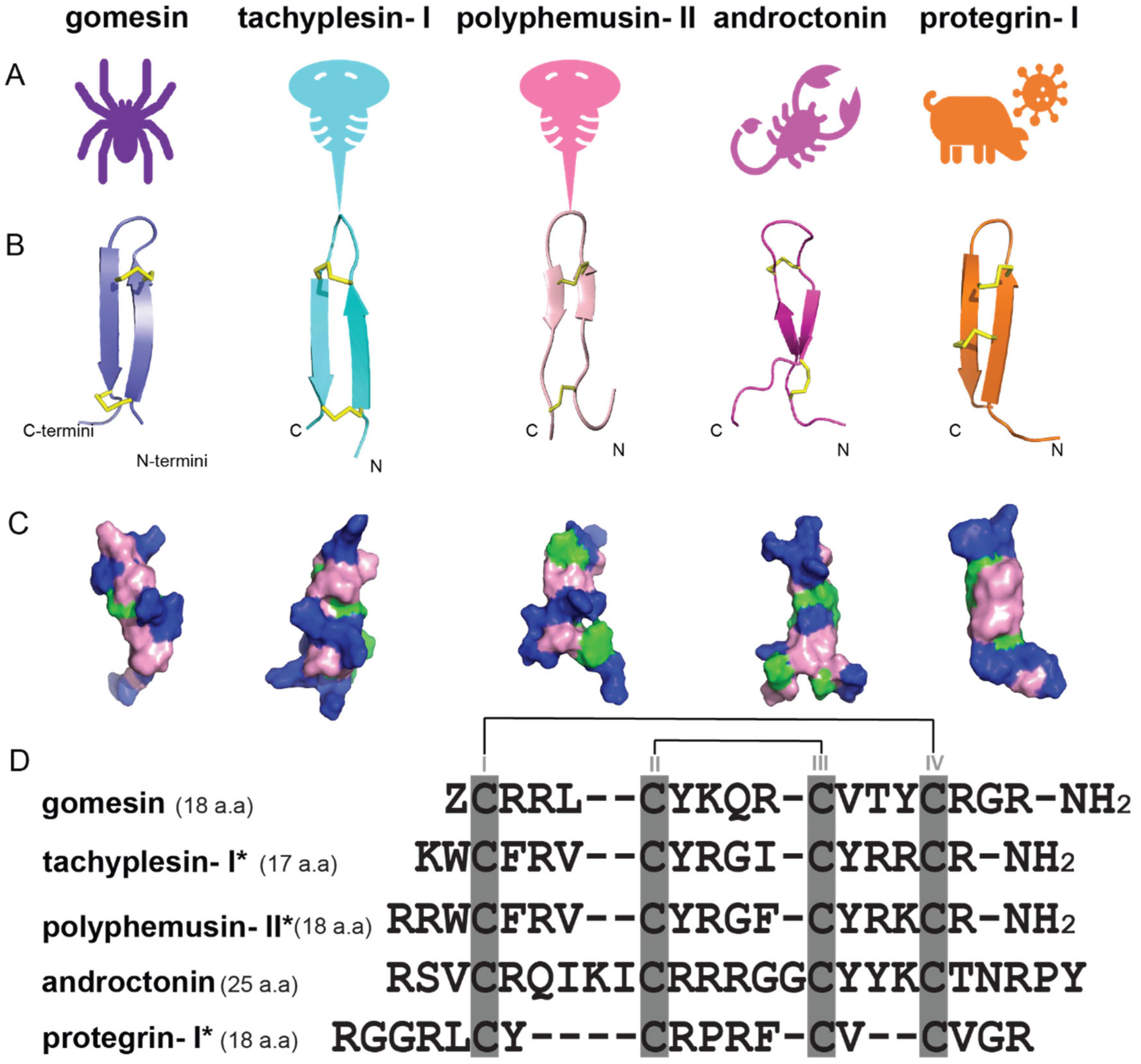
Figure 4. Structures and sequence alignment of β-hairpin AMPs. (A) Natural sources from which listed peptides were first identified: gomesin was isolated from the spider Acanthoscurria gomesiana; tachyplesin-I from the horseshoe crab Tachypleus tridentatus; polyphemusin-II from the horseshoe crab Limulus polyphemus; androctonin from the scorpion Androctonus australis; protegrin-I from the porcine leukocytes (Sus domesticus). (B) Ribbon representation of the β-hairpin-like structure of Gm (PDB ID: 1KFP), tachyplesin (PDB ID: 2RTV), polyphemusin (PDB ID: 1RKK), androctonin (PDB ID: 1CZ6), and protegrin (PDB ID: 1PG1). Disulfide bonds are represented as yellow sticks. (C) Surface representations of Gm, tachyplesin-Ⅰ, polyphemusin-II, androctonin, and protegrin-Ⅰ. Non-polar (hydrophobic), polar, and positively charged residues are shown in green, pink, and blue, respectively. (D) Sequence alignment and total amino acid (a.a) content of Gm, tachyplesin-Ⅰ, polyphemusin-II, androctonin, and protegrin-Ⅰ. Cysteine residues are highlighted by a grey box, and disulfide bond connectivities (Cys I-IV, and Cys II-III) are shown in thick black lines. *: There is more than one variant of this peptide [35,36,37,38,39,40][35][36][37][38][39][40].
The procedures for determining the structures of gomesin [23,26][23][26] and related peptides are shown in Figure 5. For gomesin, capillary zone electrophoresis was used to separate peptides in a haemolymph extract, and masses were confirmed by MALDI-TOF mass spectrometry (Figure 5A,B). Enzyme digestion and alkylation steps were performed on purified peptides followed by amino acid sequencing for sequence confirmation (Figure 5C–E). Two-dimensional (2D) proton NMR (Figure 5F,G) was used to confirm the peptide sequence and 3D overall structure. It was found that the global fold of gomesin is made up of two antiparallel β-strands joined by a four-residue non-canonical turn (Figure 5B). The β-strands are connected by two interchain disulfide bonds, and six interchain skeleton-framework hydrogen bonds further stabilise the overall structure [41]. Both disulfide bonds adopt a right-handed conformation with a twist angle close to the low-energy conformation found in disulfide bonds bridging antiparallel β-strands, which is similar to other antimicrobial peptides [27].
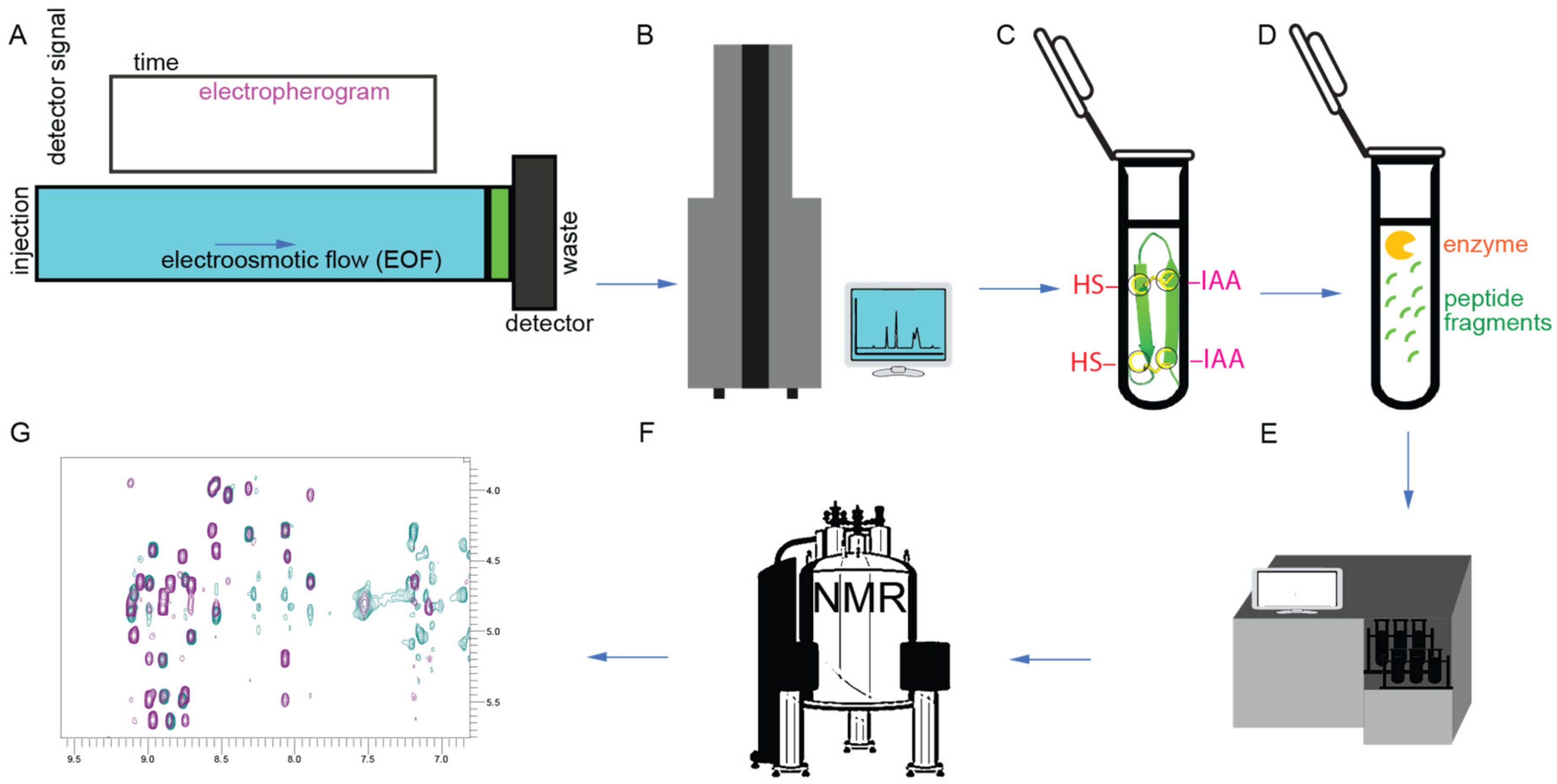
Figure 5. Structural characterization workflow. (A) Capillary zone electrophoresis. (B) MALDI-TOF-MS and electrospray ionization mass spectrometry. (C) Reduction and alkylation steps. -HS (thiol groups; dithiothreitol was used to reduce disulfide bonds) and -IAA (iodoacetamide was used for alkylation). (D) Trypsin digestion step. (E) Amino acid sequencing. (F) NMR spectrometer. (G) NMR two-dimensional (2D) spectrum [23,26][23][26].
Detailed analyses of the disulfide bond geometries, β-hairpin geometries, charges, and bioactivities for a selection of β-hairpin peptides are given in Table 1. The Cysα-Cysα distances across the two disulfide bonds in gomesin are 0.38 ± 0.10 nm (Cys6-Cys11) and 0.37 ± 0.10 nm (Cys2-Cys15). These distances are much shorter than typical Cα-Cα distances connecting other secondary structure motifs or β-sheet disulfide bonds, which is more than 0.45 nm, as found in larger, more spherical peptides/proteins. This short Cysα-Cysα distance is thus a useful marker for high stability.
Table 1.
Structural properties of antimicrobial peptides with β-hairpin-like structure.
| Antimicrobial Peptides | Function/Activity | Disulfide Connectivity |
Cysα-Cysα Distances (nm) |
Types of β-Sheets | Total Net Charge |
|---|---|---|---|---|---|
| gomesin | Antimicrobial, anticancer | Cys2-Cys15, Cys6-Cys11 |
0.37 ± 0.10, 0.38 ± 0.10 (<0.45) [41] |
right-handed rotamer, antiparallel β-sheets | +6 |
| tachyplesin-I | Antimicrobial, antifungal, and anticancer | Cys3-Cys16, Cys7-Cys12 |
0.43 ± 0.20, 0.39 ± 0.20 [40] |
right-handed rotamer, antiparallel β-sheets | +6 |
| polyphemusin-II | Antimicrobial and antifungal | Cys4-Cys17, Cys8-Cys13 |
0.43 ± 0.50, 0.37 ± 0.10 [40] |
right-handed rotamer, antiparallel β-sheets | +7 |
| androctonin | Inhibits the growth of Gram-positive bacteria | Cys4-Cys20, Cys10-Cys13 |
n.a. | right-handed rotamer, antiparallel β-sheets | +8 |
| protegrins-I | Antimicrobial and antifungal | Cys6-Cys15, Cys8-Cys13 |
0.38 ± 0.30, 0.35 ± 0.10 [40] |
right-handed rotamer, antiparallel β-sheets | +6 |
n.a., not available; Cys: cysteine.
The amphiphilic nature of Gm is another feature commonly shared by other AMP/anticancer peptides. The stable β-hairpin-like structure of Gm, as well as its positively charged and amphiphilic properties, make it highly potent with selectivity to certain cancer cell types, such as melanoma or chronic myeloid leukaemia cells [42]. Based on these findings and the proximity of the termini in gomesin, cyclic gomesin (cGm) has been used as a scaffold for drug design. It comprises a cyclic version of the native Gm with a backbone joined at the N- and C- terminus via the addition of a glycine linker residue [31]. Its structure, shown in Figure 6, has been determined and the cyclic peptide has been shown to be more stable than its linear counterpart. It enters cancer cells via a mechanism modulated by electrostatic interactions between its positively charged surface exposed residues [42[42][43],43], illustrated in Figure 6, and the negatively charged phospholipids at the outer layer of cancer cell membranes.
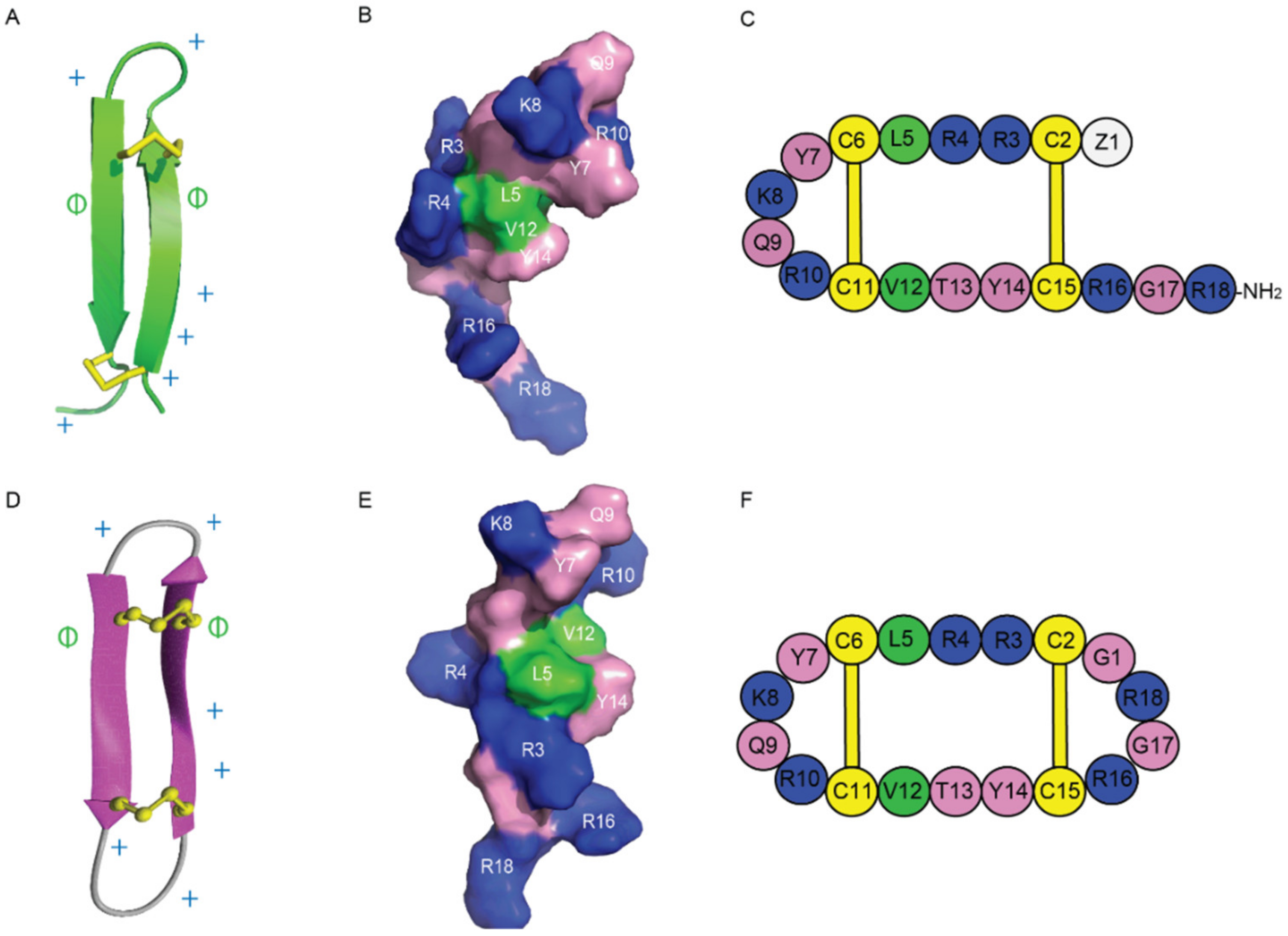
Figure 6. Structures of Gm and cGm. (A) Ribbon representation of the β-hairpin structure of Gm with disulfide bonds highlighted in yellow. PDB structure ID: 1KFP. (B) Surface representation of Gm. Non-polar (hydrophobic) residues are shown in green, polar residues in pink, and positively charged residues in blue. (C) A schematic view of Gm sequence. (D) Three-dimensional structure of cGm. Disulfide bonds are represented with yellow ball and sticks, and two antiparallel β-sheets are shown in purple. BMRB ID: 17986. (E) Surface representation of cGm. Non-polar (hydrophobic) residues are shown in green, polar residues in pink, and positively charged residues in blue. (F) A schematic view of cGm sequence. Positively charged residues are indicated with a ‘+’ symbol, hydrophobic residues are indicated with a ‘ɸ’ symbol on (A,D).
References
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655.
- WHO. Antimicrobial Resistance November 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 9 November 2022).
- CDC. About Antibiotic Resistance December 2021. Available online: https://www.cdc.gov/drugresistance/about.html (accessed on 9 November 2022).
- Khan, S.N.; Khan, A.U. Breaking the Spell: Combating Multidrug Resistant ‘Superbugs’. Front. Microbiol. 2016, 7, 174.
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202.
- Mattiuzzo, M.; Bandiera, A.; Gennaro, R.; Benincasa, M.; Pacor, S.; Antcheva, N.; Scocchi, M. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 2007, 66, 151–163.
- Mardirossian, M.; Grzela, R.; Giglione, C.; Meinnel, T.; Gennaro, R.; Mergaert, P.; Scocchi, M. The host antimicrobial peptide Bac71-35 binds to bacterial ribosomal proteins and inhibits protein synthesis. Chem. Biol. 2014, 21, 1639–1647.
- Le, C.F.; Gudimella, R.; Razali, R.; Manikam, R.; Sekaran, S.D. Transcriptome analysis of Streptococcus pneumoniae treated with the designed antimicrobial peptides, DM3. Sci. Rep. 2016, 6, 26828.
- Kragol, G.; Lovas, S.; Varadi, G.; Condie, B.A.; Hoffmann, R.; Otvos, L., Jr. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 2001, 40, 3016–3026.
- Le, C.F.; Fang, C.M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16.
- Wronska, A.K.; Bogus, M.I. Heat shock proteins (HSP 90, 70, 60, and 27) in Galleria mellonella (Lepidoptera) hemolymph are affected by infection with Conidiobolus coronatus (Entomophthorales). PLoS ONE 2020, 15, e0228556.
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656.
- Cabib, E. Two novel techniques for determination of polysaccharide cross-links show that Crh1p and Crh2p attach chitin to both beta(1-6)- and beta(1-3)glucan in the Saccharomyces cerevisiae cell wall. Eukaryot Cell 2009, 8, 1626–1636.
- Spohn, R.; Daruka, L.; Lazar, V.; Martins, A.; Vidovics, F.; Grezal, G.; Mehi, O.; Kintses, B.; Szamel, M.; Jangir, P.K.; et al. Integrated evolutionary analysis reveals antimicrobial peptides with limited resistance. Nat. Commun. 2019, 10, 4538.
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A. Antifungal peptides: To be or not to be membrane active. Biochimie 2016, 130, 132–145.
- Shu, G.; Chen, Y.; Liu, T.; Ren, S.; Kong, Y. Antimicrobial Peptide Cathelicidin-BF Inhibits Platelet Aggregation by Blocking Protease-Activated Receptor 4. Int. J. Peptide Res. Ther. 2019, 25, 349–358.
- Perlikowska, R.; Silva, J.; Alves, C.; Susano, P.; Pedrosa, R. The Therapeutic Potential of Naturally Occurring Peptides in Counteracting SH-SY5Y Cells Injury. Int. J. Mol. Sci. 2022, 23, 11778.
- Miethke, M.; Pieroni, M.; Weber, T.; Bronstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749.
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779.
- Duwadi, D.; Shrestha, A.; Yilma, B.; Kozlovski, I.; Sa-Eed, M.; Dahal, N.; Jukosky, J. Identification and screening of potent antimicrobial peptides in arthropod genomes. Peptides 2018, 103, 26–30.
- Lofgren, S.E.; Miletti, L.C.; Steindel, M.; Bachere, E.; Barracco, M.A. Trypanocidal and leishmanicidal activities of different antimicrobial peptides (AMPs) isolated from aquatic animals. Exp. Parasitol. 2008, 118, 197–202.
- Wu, Q.; Patocka, J.; Kuca, K. Insect Antimicrobial Peptides, a Mini Review. Toxins 2018, 10, 461.
- Silva, P.I., Jr.; Daffre, S.; Bulet, P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J. Biol. Chem. 2000, 275, 33464–33470.
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11, 87.
- De la torre, B.; Albericio, F. Peptide Therapeutics 2.0. Molecules 2020, 25, 2293.
- Mandard, N.; Bulet, P.; Caille, A.; Daffre, S.; Vovelle, F. The solution structure of gomesin, an antimicrobial cysteine-rich peptide from the spider. Eur. J. Biochem. 2002, 269, 1190–1198.
- Fazio, M.A.; Oliveira, V.X., Jr.; Bulet, P.; Miranda, M.T.; Daffre, S.; Miranda, A. Structure-activity relationship studies of gomesin: Importance of the disulfide bridges for conformation, bioactivities, and serum stability. Biopolymers 2006, 84, 205–218.
- Chalker, J.M.; Davis, B.G. Chemical mutagenesis: Selective post-expression interconversion of protein amino acid residues. Curr. Opin. Chem. Biol. 2010, 14, 781–789.
- Chalker, J.M.; Bernardes, G.J.; Lin, Y.A.; Davis, B.G. Chemical modification of proteins at cysteine: Opportunities in chemistry and biology. Chem. Asian J. 2009, 4, 630–640.
- Malins, L.R. Peptide modification and cyclization via transition-metal catalysis. Curr. Opin. Chem. Biol. 2018, 46, 25–32.
- Chan, L.Y.; Zhang, V.M.; Huang, Y.H.; Waters, N.C.; Bansal, P.S.; Craik, D.J.; Daly, N.L. Cyclization of the antimicrobial peptide gomesin with native chemical ligation: Influences on stability and bioactivity. ChemBioChem 2013, 14, 617–624.
- Miranda, A.; Jouvensal, L.; Vovelle, F.; Bulet, P.; Daffre, S. A powerful antimicrobial peptide isolated from the Brazilian tarantula spider Acanthoscurria gomesiana. In Animal Toxins: State of the Art. Perspectives in Halth and Biotechnology; de Lima, M.E., de Castro Pimenta, A.M., Martin-Eauclaire, M.F., Zingali, R.B., Rochat, H., Eds.; UFMG: Belo Horizonte, Brazil, 2009; pp. 227–248.
- Chan, W.; White, P. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: Oxford, UK, 1999.
- Machado, A.; Fazio, M.A.; Miranda, A.; Daffre, S.; Machini, M.T. Synthesis and properties of cyclic gomesin and analogues. J. Pept. Sci. 2012, 18, 588–598.
- Kawano, K.; Yoneya, T.; Miyata, T.; Yoshikawa, K.; Tokunaga, F.; Terada, Y.; Iwanaga, S. Antimicrobial peptide, tachyplesin I, isolated from hemocytes of the horseshoe crab (Tachypleus tridentatus). NMR determination of the beta-sheet structure. J. Biol. Chem. 1990, 265, 15365–15367.
- Nakamura, T.; Furunaka, H.; Miyata, T.; Tokunaga, F.; Muta, T.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Tachyplesin, a class of antimicrobial peptide from the hemocytes of the horseshoe crab (Tachypleus tridentatus). Isolation and chemical structure. J. Biol. Chem. 1988, 263, 16709–16713.
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin II, and polyphemusins I and II: Chemical structures and biological activity. J. Biochem. 1989, 106, 663–668.
- Mandard, N.; Sy, D.; Maufrais, C.; Bonmatin, J.M.; Bulet, P.; Hetru, C.; Vovelle, F. Androctonin, a novel antimicrobial peptide from scorpion Androctonus australis: Solution structure and molecular dynamics simulations in the presence of a lipid monolayer. J. Biomol. Struct. Dyn. 1999, 17, 367–380.
- Kokryakov, V.N.; Harwig, S.S.; Panyutich, E.A.; Shevchenko, A.A.; Aleshina, G.M.; Shamova, O.V.; Korneva, H.A.; Lehrer, R.I. Protegrins: Leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett. 1993, 327, 231–236.
- Deplazes, E.; Chin, Y.K.; King, G.F.; Mancera, R.L. The unusual conformation of cross-strand disulfide bonds is critical to the stability of beta-hairpin peptides. Proteins 2020, 88, 485–502.
- Tanner, J.D.; Deplazes, E.; Mancera, R.L. The Biological and Biophysical Properties of the Spider Peptide Gomesin. Molecules 2018, 23, 1733.
- Troeira Henriques, S.; Lawrence, N.; Chaousis, S.; Ravipati, A.S.; Cheneval, O.; Benfield, A.H.; Elliott, A.G.; Kavanagh, A.M.; Cooper, M.A.; Chan, L.Y.; et al. Redesigned Spider Peptide with Improved Antimicrobial and Anticancer Properties. ACS Chem. Biol. 2017, 12, 2324–2334.
- Benfield, A.H.; Defaus, S.; Lawrence, N.; Chaousis, S.; Condon, N.; Cheneval, O.; Huang, Y.H.; Chan, L.Y.; Andreu, D.; Craik, D.J.; et al. Cyclic gomesin, a stable redesigned spider peptide able to enter cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183480.
More