Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Mariusz Mojzych.
Sharpless asymmetric dihydroxylation is an important reaction in the enantioselective synthesis of chiral vicinal diols that involves the treatment of alkene with osmium tetroxide along with optically active quinine ligand. Sharpless introduced this methodology after considering the importance of enantioselectivity in the total synthesis of medicinally important compounds.
- Sharpless asymmetric dihydroxylation
- natural products
- enantioselective
1. Introduction
Asymmetric synthesis plays an important role in the stereoisomeric and enantiomeric synthesis of drugs [1]. In this regard, Sharpless in the 1980s introduced Sharpless epoxidation, which is a facile methodology for the asymmetric synthesis of 2,3-epoxy alcohols by using primary and secondary allylic alcohols [2]. Considering the importance of enantioselective synthesis of Sharpless epoxidation, Sharpless asymmetric dihydroxylation was then employed by Sharpless to synthesize vicinal diols [3,4][3][4]. In this methodology, chiral vicinal diol moiety is obtained by the reaction of an alkene with osmium tetroxide in the presence of an optically active quinine ligand [5,6][5][6].
The presence of a chiral ligand is the primary requirement for osmium catalyzed bishydroxylation [7]. Since the inception of this protocol, various co-oxidant ligands have been employed by different researchers. However, it was inferred that low yields of diol were obtained by employing stoichiometric oxidants, i.e., H2O2 and NaClO3 or KClO3 [8,9][8][9]. Improvements in the yield of target molecules were observed by employing Upjohn dihydroxylation in the presence of N-methylmorpholine N-oxide and by using alkaline tert-butyl hydroperoxide (t-BHP) [10]. Moreover, potassium hexacyanoferrate (III) has been found to be the most effective in recent times. Similarly, Sharpless also utilized this optically active terminal oxidant for the bishydroxylation of alkenes [11]. As time passed, Sharpless and his coworkers devoted their attention to the use of cinchona alkaloids as chiral ligands, which gave tremendous yields with high enantioselectivity. Later on, pyrimidine and phthalazine incorporated dimeric alkaloids were employed as asymmetric ligands for the efficient synthesis of vicinal diols. K2OsO2(OH)4, K2CO3, (DHQD)2PHAL, (DHQ)2PHAL, and K3Fe(CN)6 were found to be mandatory chemical substances for carrying out Sharpless asymmetric dihydroxylation. The mixture of these four reagents is referred to as “AD-mix”. The (DHQ)2PHAL-containing mixture is termed “AD-mix-α”, while “AD-mix-β” includes (DHQD)2PHAL as ligand [11,12][11][12]. The general scheme for osmium-catalyzed asymmetric dihydroxylation is given below [12] (Scheme 1).
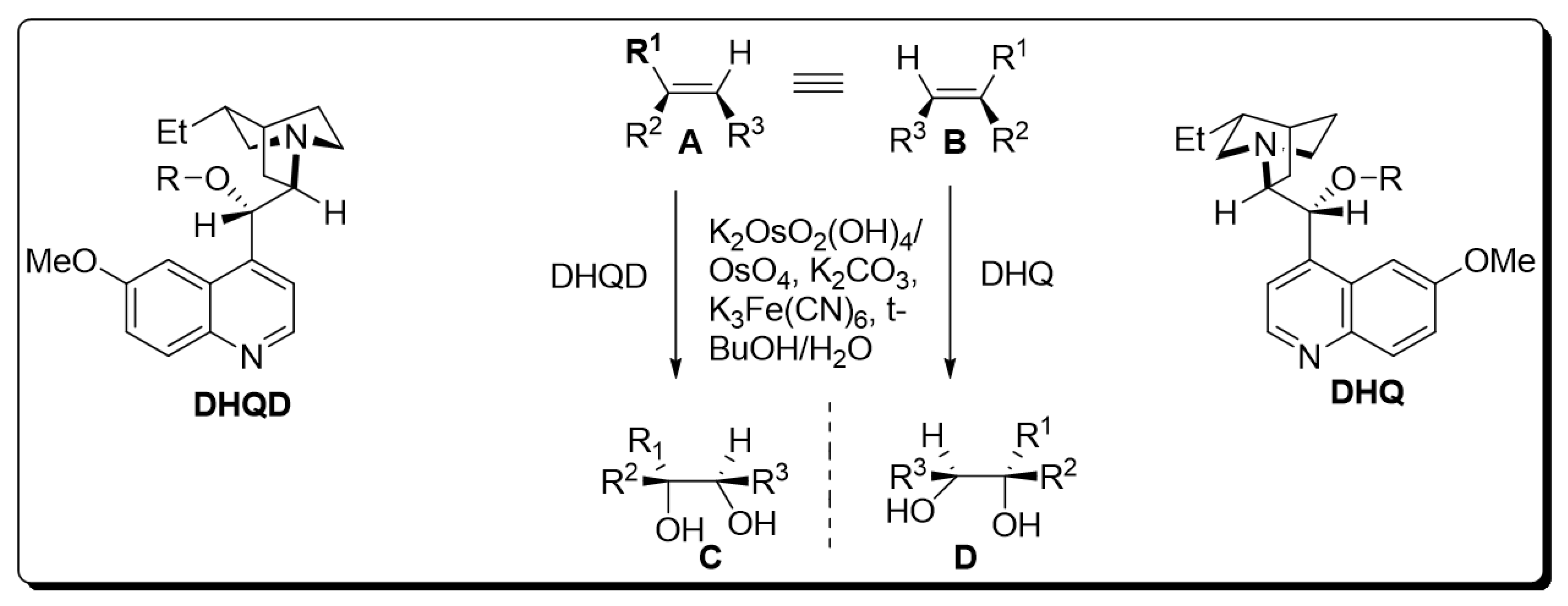
Scheme 1. Osmium-catalyzed asymmetric dihydroxylation.
Sharpless asymmetric dihydroxylation has found tremendous applications in the synthesis of a variety of naturally occurring biologically active compounds [13,14][13][14]. Total synthesis of various natural products such as alkaloids, lactones, amino acids, flavones, polyketides, macrolides, glycosides, terpenes, etc., involves Sharpless asymmetric dihydroxylation as an integral step [15,16,17,18][15][16][17][18]. This protocol results in the high enantioselectivity of vicinal diols, which are then reacted further in the presence of required reagents to synthesize the desired medicinally important target molecules [19,20][19][20]. The following figure illustrates the structure of a few biologically important natural products, whose total synthesis is achieved by employing Sharpless asymmetric dihydroxylation as an essential step (Figure 1) [21,22][21][22].
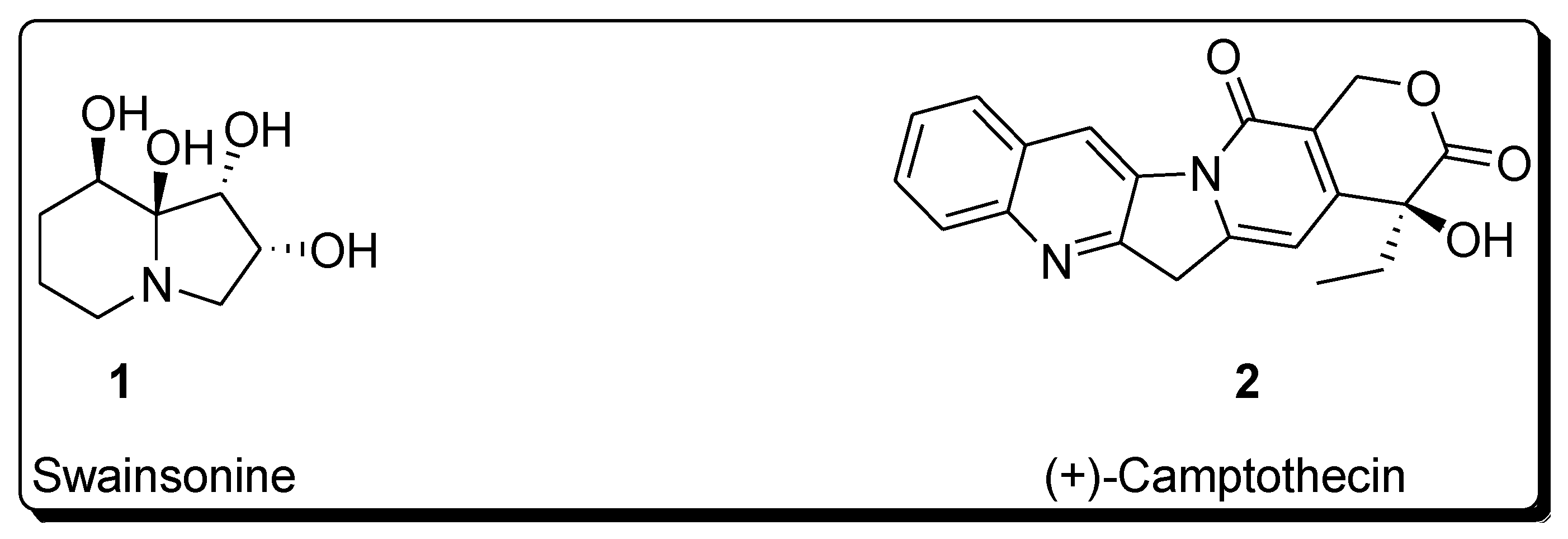
Figure 1. Structure of biologically important organic compounds employing Sharpless asymmetric dihydroxylation in their total synthesis.
2. Synthesis of Alkaloid-Based Natural Products
2.1. Lycorine-Type Alkaloids
Zephyranthine belongs to the class of lycorine alkaloids, which are known for their medicinal usage [26][23]. Members of this class have been found to interrupt the acetylcholine activity and uncontrolled division of cancer cells [27][24]. Zhao et al. [28][25] in 2021 reported the efficient synthesis of (-)-zephyranthine by utilizing two one-pot reactions. Evans’ nickel (II) catalyst 9 initiated the synthesis by reacting unsaturated β-ketoeaster 5 and nitro olefin 8 via a series of Michael addition reactions. Both reactants 5 and 8 were prepared individually. In this regard, starting material 2,2,6-trimethyl-[1,3][1][3]dioxin-4-one 3 was reacted with diethylchlorophosphite and LiHMDS followed by oxidation in the presence of hydrogen peroxide, thereby giving compound 4 in 90% yield, which was then reacted with methanol and sodium hydride in the presence of tetrahydrofuran to give compound 5 in 87% yield. Then, substituted carbaldehyde 6 was reacted with p-TSA and ethylene glycol followed by treatment with toluene and diethylether, which resulted in the synthesis of compound 7 in 90% yield. The compound 7 was then reacted with MeNO2 and trifluoroacetic acid to obtain compound 8 in 84% yield. For the first Michael addition, use of toluene and Triton B were considered to be effective after optimizing the various reaction conditions. This reaction resulted in 85% yield of penta-substituted cyclohexane 10, which is highly enantioselective (90% ee) and (>20:1) diastereoselective in nature. In order to prevent the undesirable aldol reaction, the enol moiety in 10 was protected by treating with benzoylchloride (BzCl) in the presence of pyridine, which resulted in 97% yield of benzoate 11 with more than 99% enantioselectivity. The compound 11 was cyclized through one-pot reaction on treatment with HBr and acetic acid in the presence of tetrahydrofuran followed by reaction with zinc powder and hydrochloric acid, obtaining intermediate 12 in 57% yield. The resulting intermediate was treated with Comins’ reagent and LiHMDS in the presence of tetrahydrofuran, resulting in the synthesis of triflate, which was further treated with palladium acetate, formic acid and triphenylphosphine to synthesize compound 13 in 85% yield. In the last step, enantioselective synthesis of zephyranthine 14 in quantitative yield (67%) with 7.2:1 diastereoselectivity ratio was achieved by Sharpless asymmetric dihydroxylation on reaction with AD-mix-β in the presence of tetrahydrofuran or water (Scheme 2).
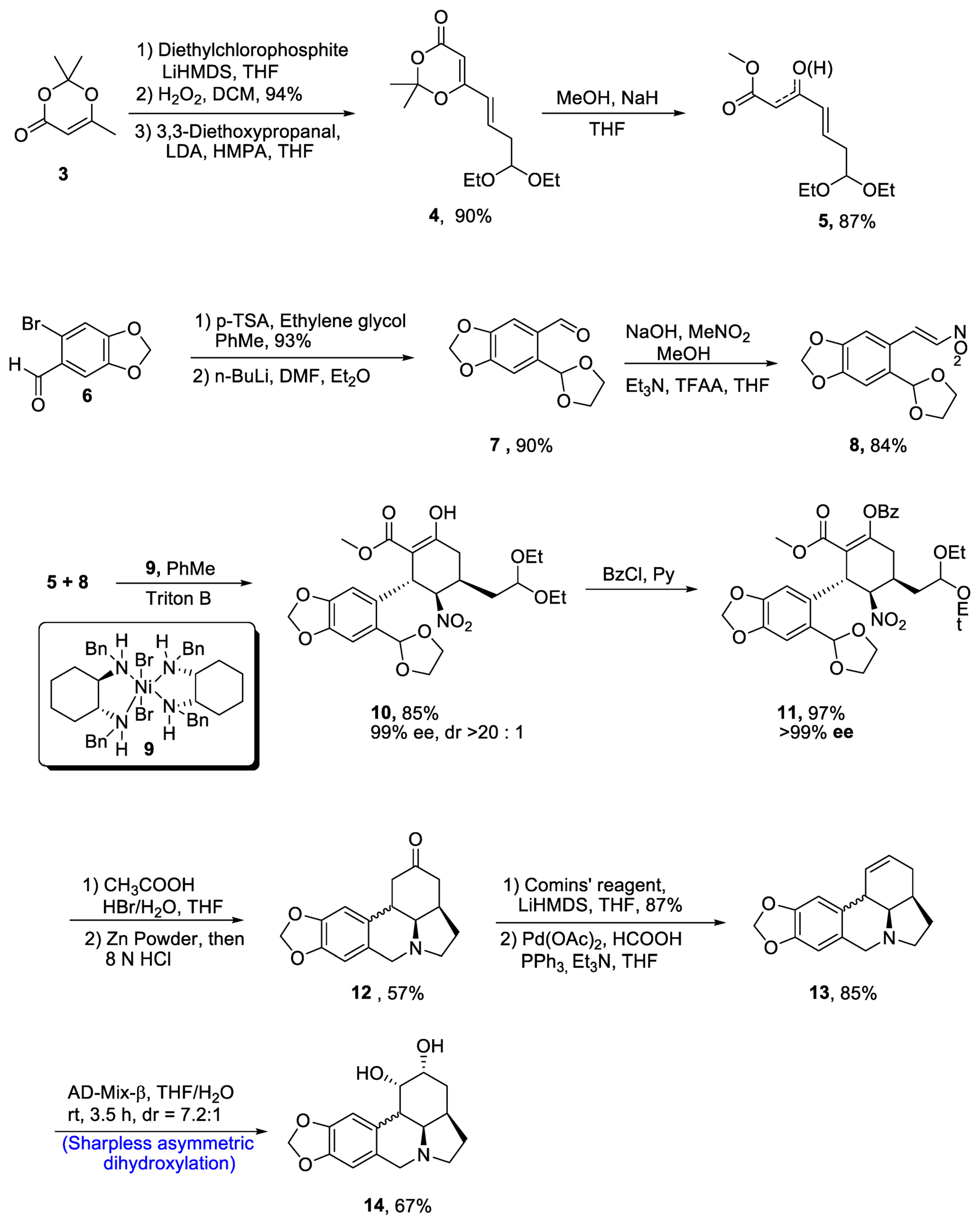
Scheme 2. Synthesis of (−)-zephyranthine 14.
2.2. Muscarine Alkaloid
There are several tetrahydrofurans containing natural products including various alkaloids such as muscarine, epi-muscarine and allo-muscarine, which are extracted from Amanita muscaria, i.e., a mushroom species [29][26]. Muscarine and its derivatives are highly potent pharmacological agents that also act as acetylcholine inhibitors [30][27]. Owing to their wide biological importance, Gehlawat et al. [31][28] in 2020 reported the total synthesis of epi-mucarine alkaloid by using various reactions including Sharpless asymmetric dihydroxylation, cyclization by bimolecular nucleophilic substitution reaction, and cleavage of epoxide ring. In the first step, (p-methoxybenzyl) glycidyl ether 15 was treated with propyl lithium in the presence of boron triflouride diethyl etherate followed by reduction with lithium aluminum hydride, resulting in the synthesis of compound 16 in 86% yield. The compound 16 was further treated with tosyl chloride in the presence of dimethyl aminopyridine and triethylamine followed by reaction with osmium tetroxide and potassium hexacyanoferrate (III) via Sharpless asymmetric dihydroxylation to obtain tosyl protected diol intermediate 17. This newly synthesized intermediate was subjected to base catalyzed reaction to prepare compound 18 in 90% yield. Compound 18 was further made to react with cerium ammonium nitrate in the presence of acetonitrile followed by treatment with imidazole, pyrrolidine and toluene. Synthesis of epi-muscarine 19 was achieved by treatment with trimethyl amine and methanol in 95% yield (Scheme 3).
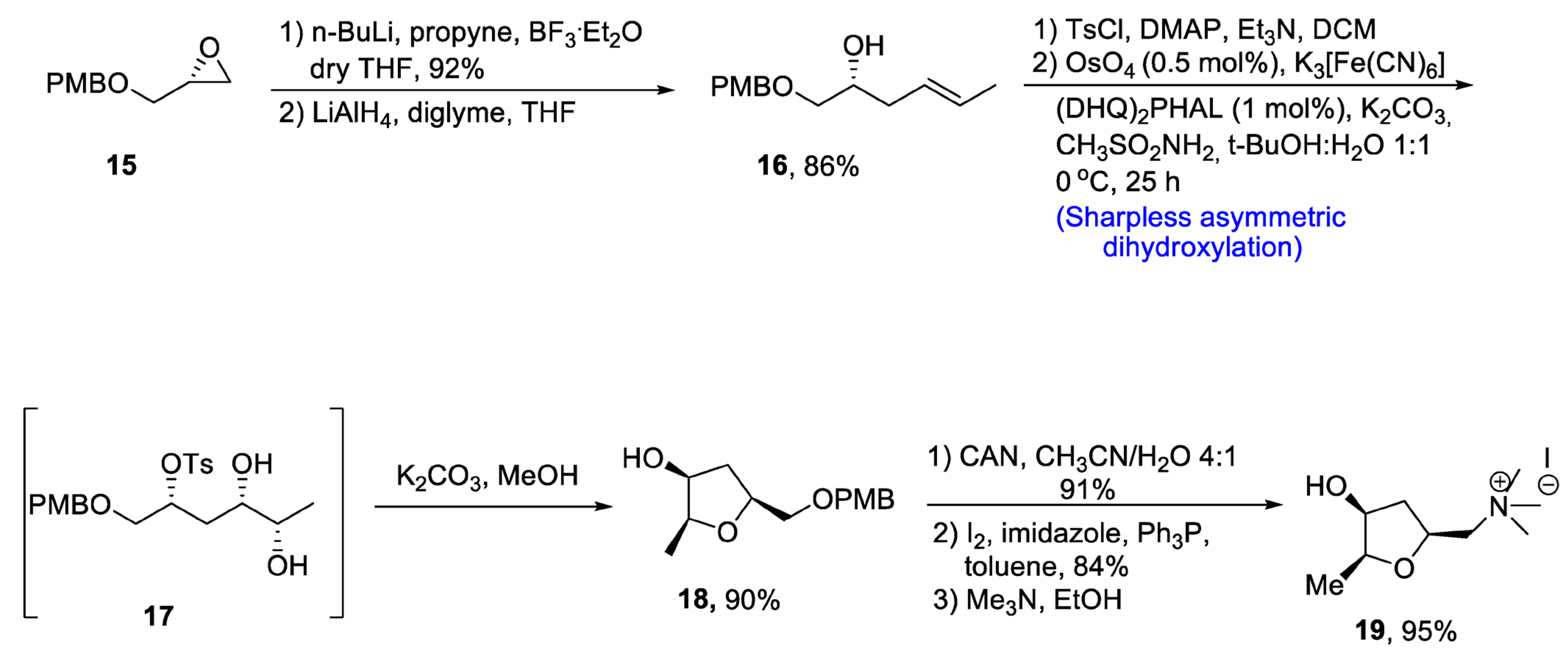
Scheme 3. Synthesis of epi-muscarine alkaloid 19.
2.3. Monoterpenoid Indole Alkaloid
A major class of natural products comprises monoterpenoid indole alkaloids (MIAs), which are effective against a wide range of bacterial, viral and neurological disorders [32][29]. These are also useful against tumor-causing agents [33,34][30][31]. Depending upon the positioning of atoms, MIAs are divided into three categories, most of which are derived from different parts of Alstonia scholaris. Zhang et al. in 2020 [35][32] proposed the total synthesis of two important MIAs, i.e., alstolarines A and B. They stated that the total synthesis could be initiated with the forerunner difforlemenine 20, which could be subjected to reduction followed by ring opening reaction, leading to the generation of compound 21. Then, nucleophilic substitution could result in the synthesis of compound 22, a common precursor in the synthesis of alstolarines A and B. For the synthesis of alstolarine A, compound 22 could be subjected to dehydration followed by Sharpless asymmetric dihydroxylation to obtain compound 23, which could synthesize target molecule 24 via cyclization of acetal. Similarly, oxidation and removal of carboxylic acid could synthesize alstolarine B 25 (Scheme 4). Both compounds were tested for their acetylcholinase inhibitory potential, and molecule 25 showed average inhibitory potential with an IC50 value = 19.3 µM.
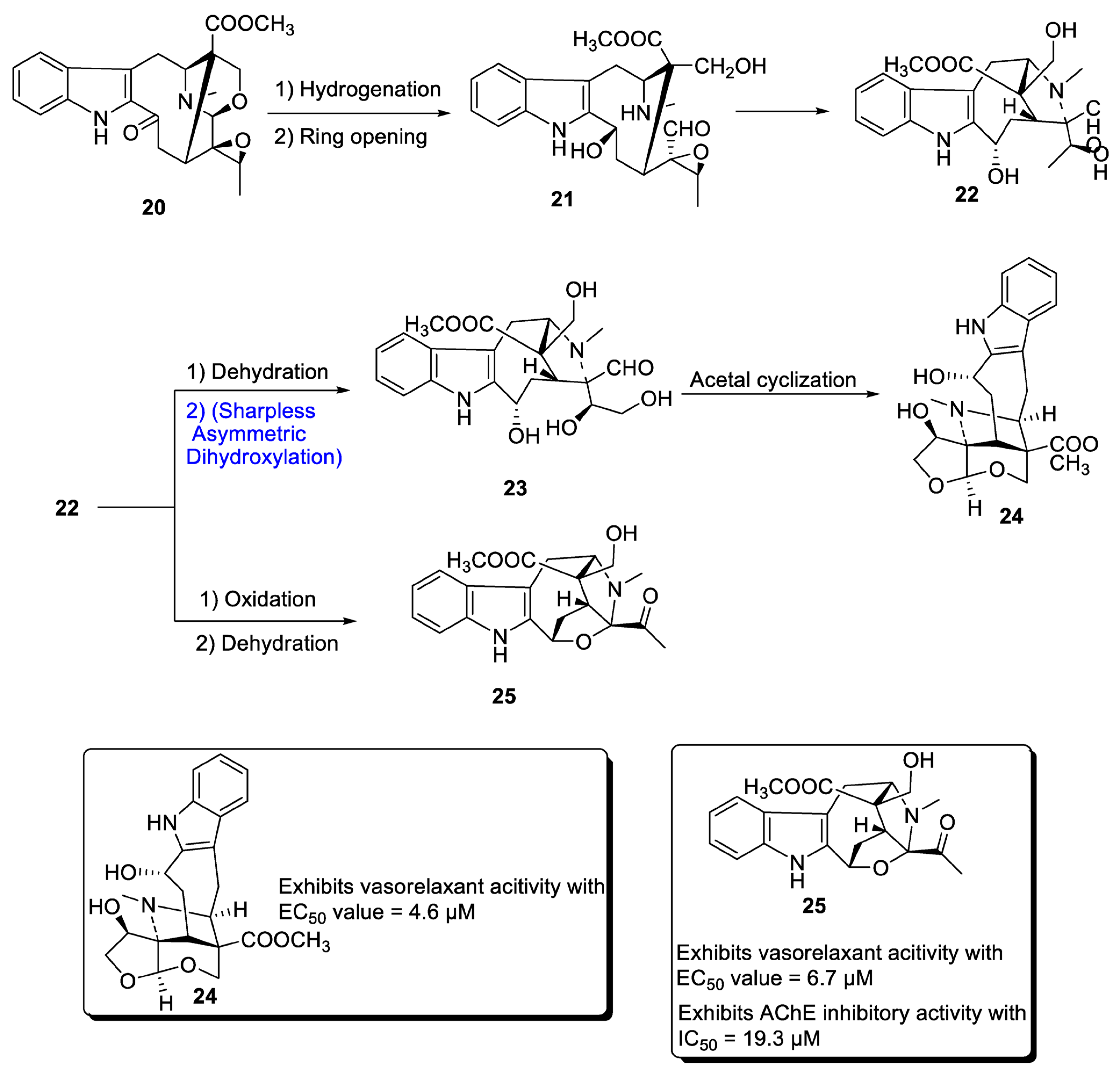
Scheme 4. Synthesis of alstolarines A 24 and B 25.
2.4. Glyphaeaside Alkaloids
The glyphaeaside alkaloids are a class of iminosugars that are found in nature, as they are derived from plants, i.e., Glyphaea brevis [36][33]. These natural products belong to the family of carbohydrates whose structures are composed of many hydroxyl groups along with phenylalkyl side-chains [37][34]. On the basis of the arrangement of he iminosugar center, the glyphaeasides have been categorized into three classes, named A, B and C. Glyphaeaside C, which was formerly referred to as 1-deoxynojirimycin, has been found to exhibit highly efficient inhibitory activity against β-glucosidase and snail β-mannosidase. However, it was determined that the inhibitory activity of glyphaeasides is dependent on the role of side chains. Moreover, the total synthesis of glyphaeaside C revealed that its structure closely resembles that of 2,5-dideoxy-2,5-imino-D-mannitol (D-DMDP). In 2021, Byatt et al. [38][35] reported the multi-step facile synthesis of glyphaeaside C. The total synthesis of ourthe target molecule initiated with the synthesis of pyrrolidine fragments from readily available starting reagent, i.e., 2,3,5-tri-O-benzyl-β-D-arabinofuranose 26. The fragments were then reacted to give oxazolidinone a and b. In the next step, commonly available 1-allly-4-methoxy-benzene 28 was treated with boron tribromide followed by reaction with benzyl bromide, leading to the protection of hydroxyl group. Then, Grubb’s first-generation catalyst was employed to treat pyrrolidine fragment 27 with benzyl group-protected 4-allylphenol, leading to the synthesis of a racemic mixture of compound 29 in 66–76% yield. Compound 29 was then subjected to α and β type Sharpless asymmetric dihydroxylation one by one, thereby giving 30 and 31 in 87% and 90% yields, respectively. Compounds 30 and 31 were further subjected to deprotection, leading to the synthesis of four diastereomers of ourthe target molecule. In the end, RP-HPLC was used to separate the glyphaeaside C (Scheme 5).
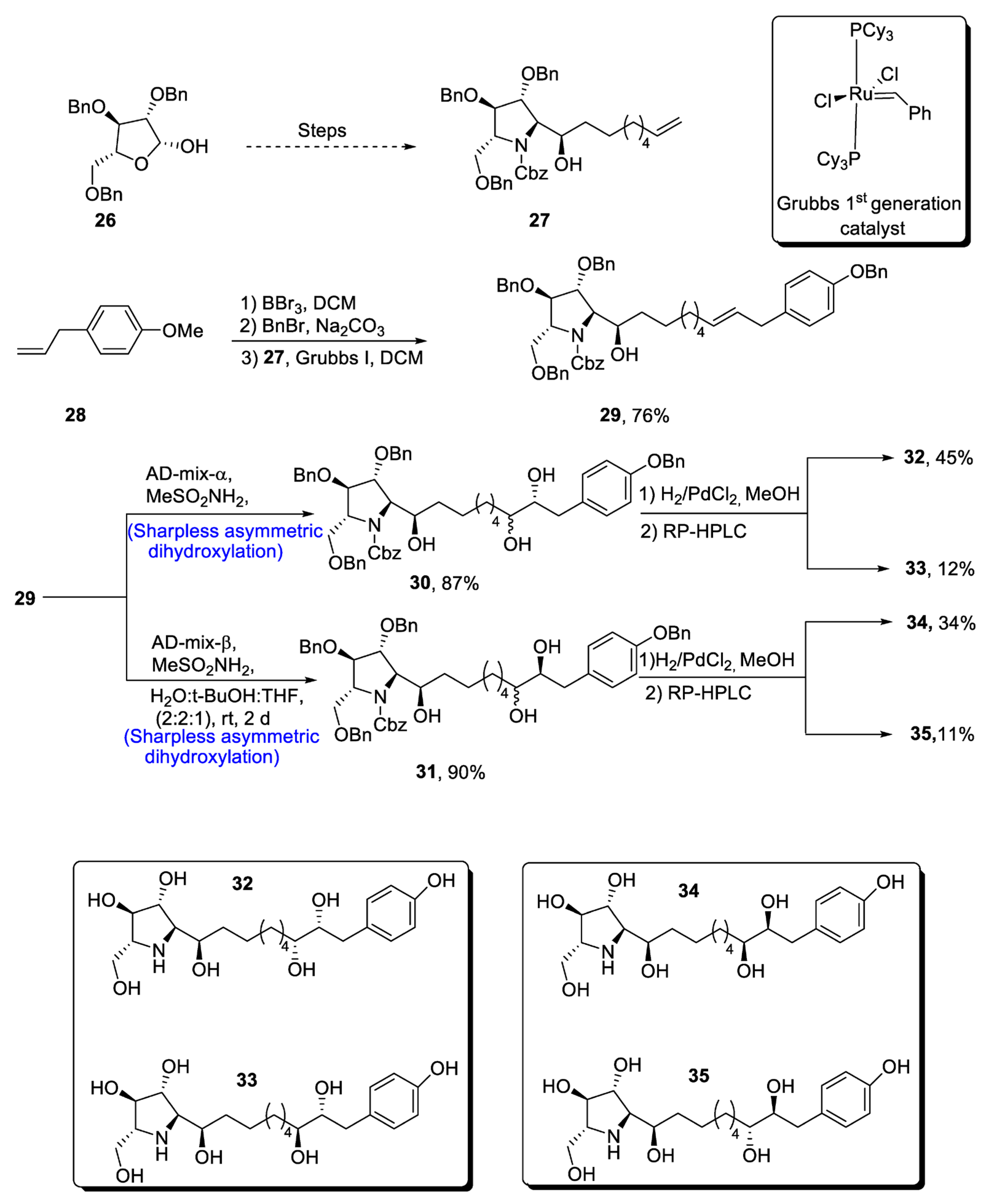
Scheme 5. Synthesis of glyphaeaside C.
3. Synthesis of Terpene-Based Natural Products
3.1. Sesquiterpenoids
Englerin is isolated from Phyllanthus engleri, and it has been found to exhibit significant cytotoxic potential against cancer cells of kidney [39][36]. Moreover, englerin A is responsible for activation of protein kinase, hence regulating the glucose level [40][37]. Its structure is composed of seven chiral carbons along with epoxyguaine framework. Considering the significance of englerin A, various researchers have attempted to report its total synthesis. In 2020, Mou et al. [41][38] described an efficient synthetic route for the total synthesis of englerin A. For this purpose, 36 was oxidized in the presence of m-chloroperoxybenzoic acid, sodium bicarbonate and dichloromethane followed by reaction with PhSiH3 in the presence of acetone and Co catalyst to synthesize compound 37 in 87% yield. Compound 37 was then subjected to Sharpless asymmetric dihydroxylation in the presence of K2OsO4, (DHQ)2PHAL and methane sulfonamide to obtain compound 38 in 59% yield. Compound 38 was further used in the synthesis of englerin A 39 (99% yield) (Scheme 6).
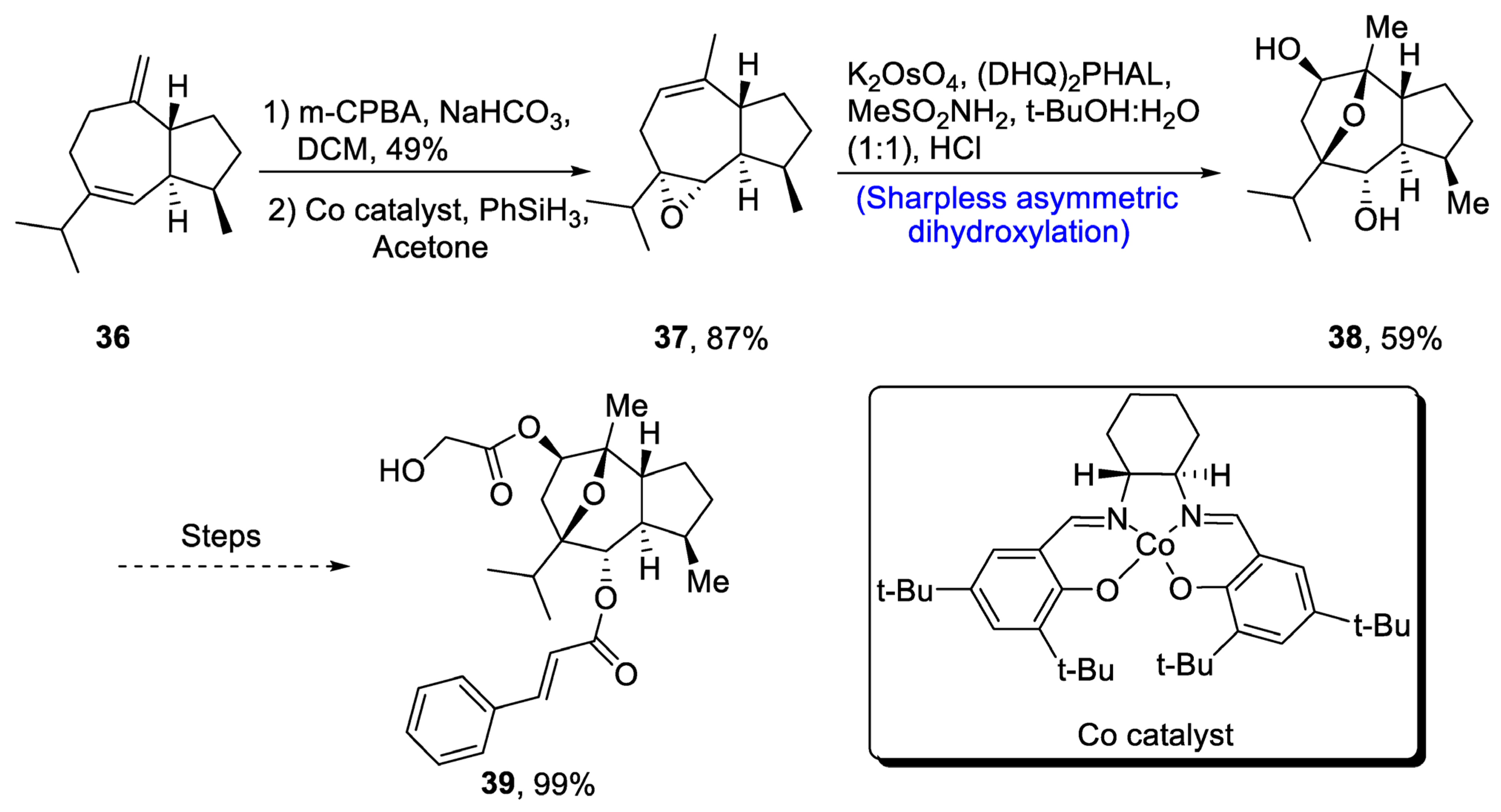
Scheme 6. Synthesis of (−)-englerin A 39.
Goyazensolide is a biologically active, naturally occurring furanoheliangolide sesquiterpenoid that plays an effective role in combating tumor cells [42][39]. It is isolated from plants, and its derivatives have been found to be more active against cancer cell lines, e.g., 15-deoxygoyazensolide [43][40]. Considering the biologically active potential of goyazensolide, Liu et al.. [44][41] in 2021 reported the total synthesis of this natural product. In the first step of the total synthesis, compound 40 was reacted with Dess–Martin periodinane followed by treatment with trimethylsilylacetylene via Sonogashira coupling, which yielded compound 41 in 72% yield. Compound 41 was then subjected to Sharpless asymmetric dihydroxylation via (DHQD)2Pyr followed by protection with tert-butyldiphenylsilyl group in the presence of imidazole and dimethylaminopyridine, which gave compound 42 in 80% yield. Compound 42 then underwent a number of steps leading to the synthesis of goyazensolide 43 in 41% yield (Scheme 7).
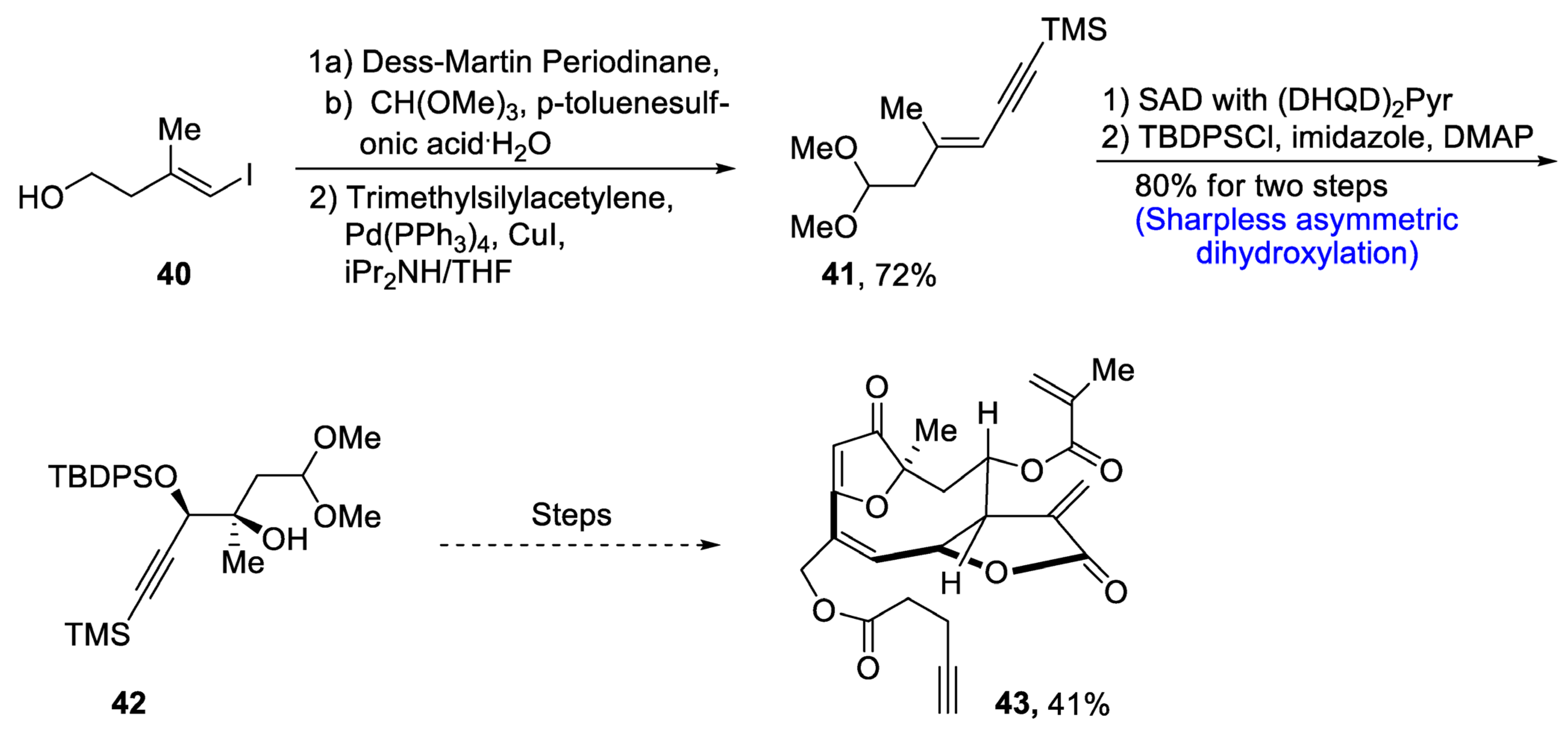
Scheme 7. Synthesis of goyazensolide 43.
Aromatic bisabolanes belong to the class of monocyclic sesquiterpenoids, and they are obtained from different living sources such as microorganisms, plants insects, etc. [45][42]. Various aromatic bisabolanes have been synthesized in their respective enantiomeric forms [46][43]. Yajima et al. [47][44] in 2021 reported the total synthesis of 1,3,5-bisabolatrien-7-ol, which involved the formation of a chiral center via Sharpless asymmetric dihydroxylation. In the first step of synthesis, compound 44 was treated with vinylmagnesium bromide followed by a reaction with methanesulfonic acid in the presence of THF and water, leading to the formation of compound 45 in 82% yield. After treatment with mercaptobenzothiazole, Sharpless asymmetric dihydroxylation was carried out, which gave chiral diol compound 47 in 98% yield. Compound 47 was further subjected to oxidation with meta-chloroperoxybenzoic acid followed by Smiles rearrangement, which gave compound 48 in 58% yield with er >99:1. Hydrogenation of compound 48 in the presence of platinum oxide led to the synthesis of S-Enantiomer 49 of 1,3,5-bisabolatrien-7-ol in 85% yield with an enantioselective ratio of more than 99:1. Similarly, R-Enantiomer 51 was achieved in an enantiomeric ratio of more than 99:1 by Sharpless asymmetric dihydroxylation of compound 46 along with similar steps (Scheme 8).
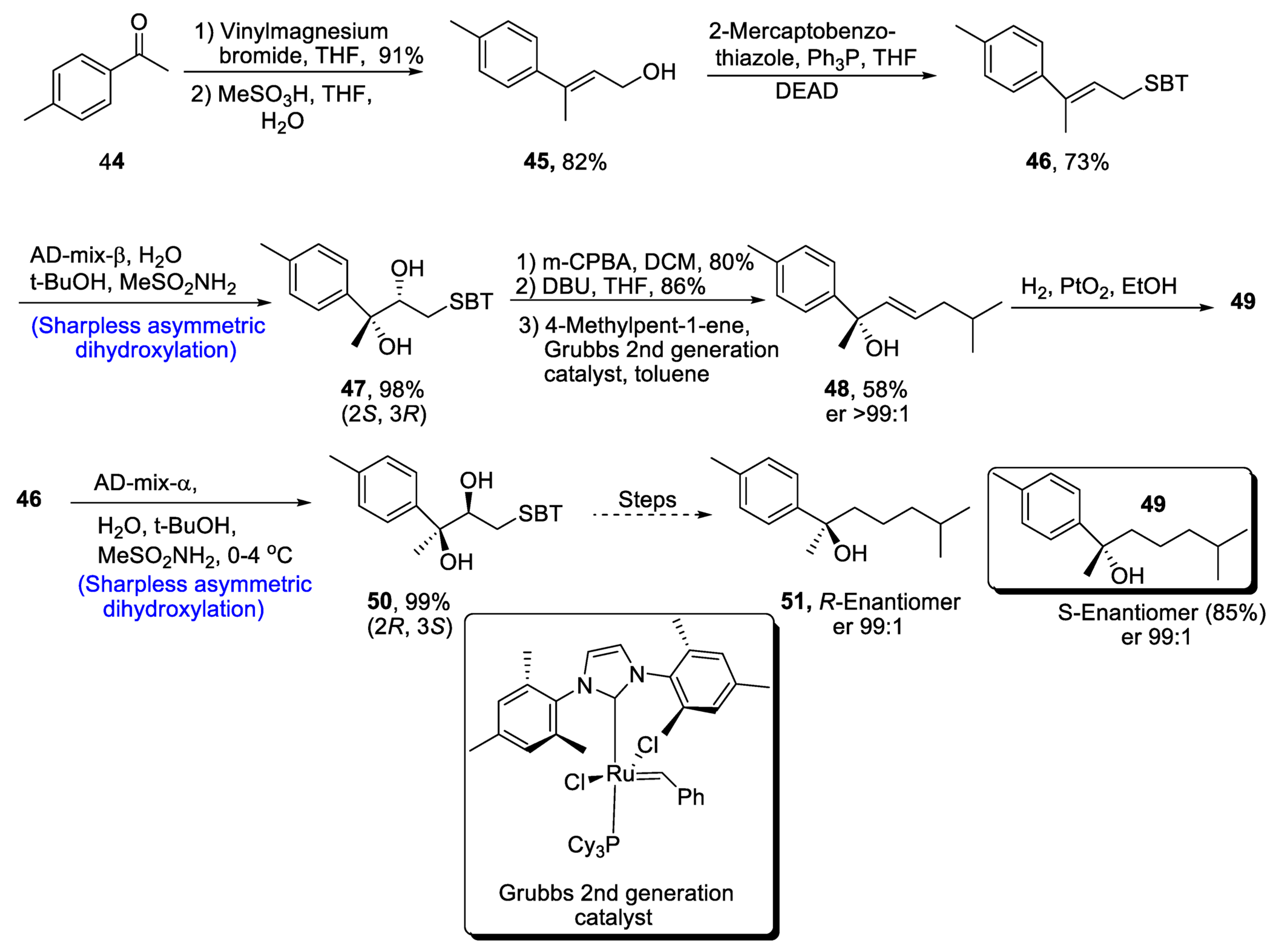
Scheme 8. Synthesis of aromatic bisabolanes 49 and 51.
Through extensive research, it has been found that the compounds isolated from the plant Phythallus engleri possess mighty anti-cancerous potential [48][45]. Owing to being composed of a glycolic ester attachment, englerin A is a more promising anti-cancerous agent as compared to englerin B, which indicates that this functional group is responsible for increased cytotoxic potential [49][46]. There have been numerous reports covering the total synthesis of englerin-A. So, in 2021, Palli et al. [50][47] attempted to explain the total synthesis of englerin A by starting with commercially available limonene 52. Limonene 52 was transformed into compound 53 by using a number of previously reported steps, followed by its treatment with methanesulfonamide, thus carrying out Sharpless asymmetric dihydroxylation to give compound 54 in 80% yield with a more than 99% diastereoselectivity ratio. Compound 54 was then subjected to protection of diol groups on treatment with 2,2-DMP and CSA followed by hydrogenation, which gave compound 55 in 82% yield. Compound 55 then underwent a number of different steps to give a mixture of oxatricyclic alcohol 56a and 56b. In the next step, compound 56 was then subjected to acylation followed by elimination in the presence of Burgess reagent to obtain compound 57 in 86% yield. Compound 57 was further used to obtain target molecule 58 in 86% yield (Scheme 9).
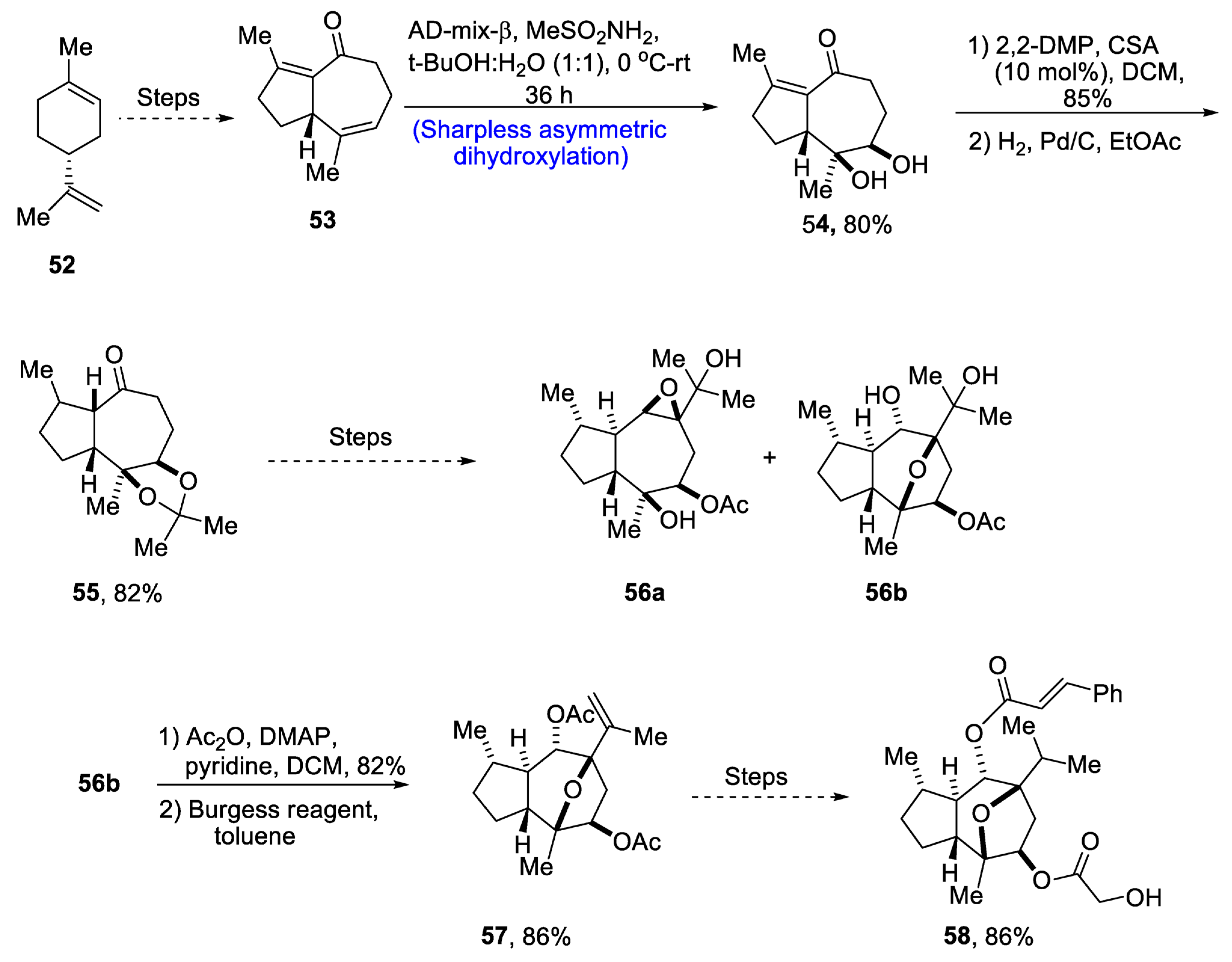
Scheme 9. Synthesis of 4-epi-englerin A 58.
3.2. Neoclerodane Diterpene
Neoclerodane diterpene, (-)-salvinorin A is derived from Salvia divinorum, which is a medicinal plant [51][48]. This diterpene is used as a medicinal agent and employed as a treatment for stress, anxiety and pain. Owing to its wide medicinal importance, various research groups have reported different routes for its synthesis. Recently, in 2021, Zimdar et al. [52][49] devised an efficient strategy for the total synthesis of salvinorin. The total synthesis was initiated with the synthesis of lactone via a number of steps resulting in the generation of lactone 60 in 87% yield. Compound 60 was further reacted with osmium tetroxide and N-methylmorpholine N-oxide in the presence of acetone and water followed by its reaction with PIDA in the presence of dichloromethane, giving keto-aldehyde 61 in 88% yield. Compound 61 further underwent a number of steps to synthesize a racemic mixture of 62 and 63 in 44% and 14% yields, respectively, with more than 99% diastereoselectivity. Compound 61 was treated with chromium trichloride and iodoform in the presence of THF to obtain compound 64 in 58% yield. Compound 64 was then further subjected to a number of steps including Mitsunobu esterification and others to obtain compound 65 in 66% yield with 94% diastereoselectivity. Compound 65 was then subjected to Sharpless asymmetric dihydroxylation, leading to the synthesis of diol moiety 66 in 95% yield. Finally, the last step involved treatment with acetic acid and DBAD(di-tert-butyl azodicarboxylate), thereby giving salvinorin A 67 in 92% yield (Scheme 10).
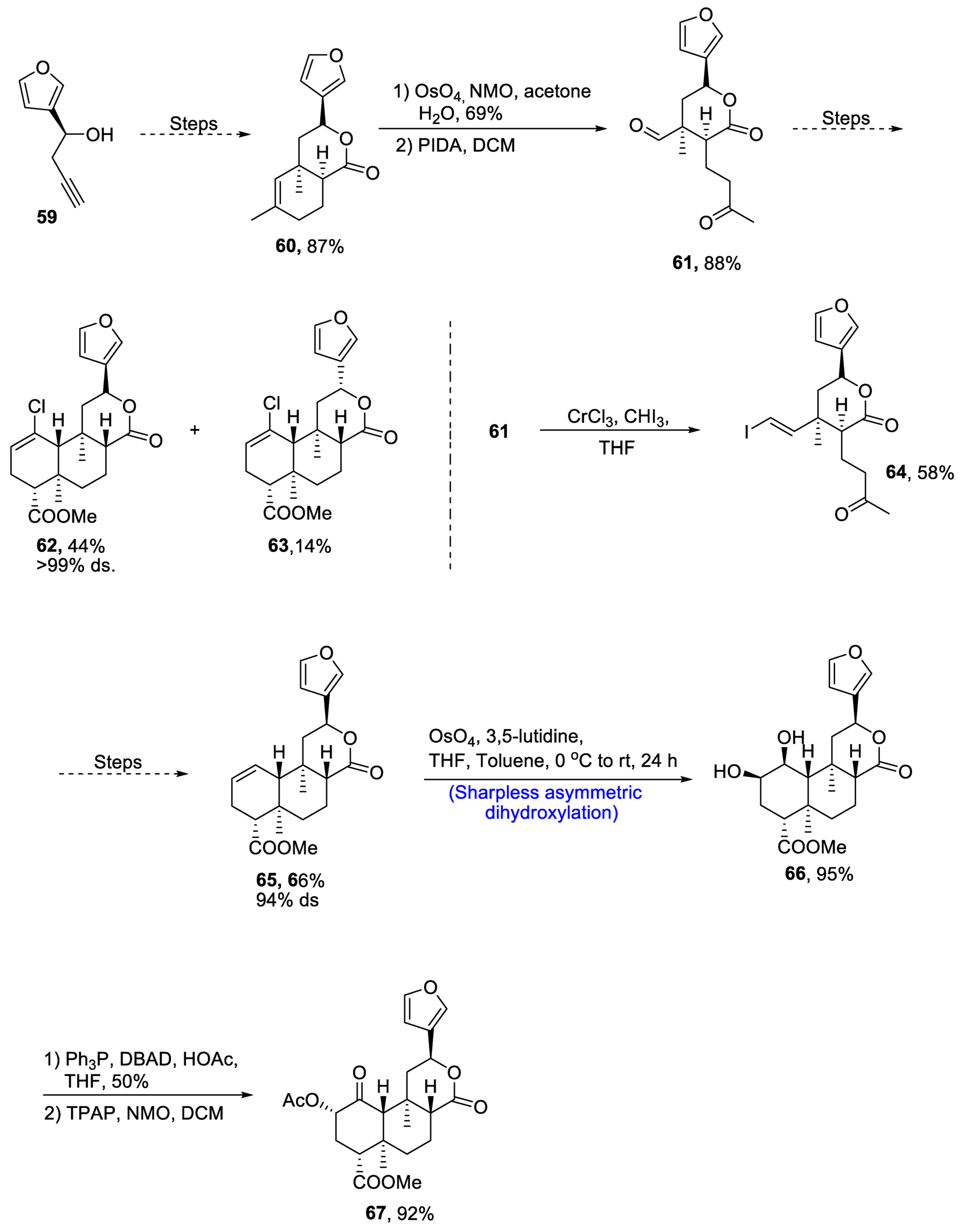
Scheme 10. Synthesis of (−)-salvinorin A 67.
3.3. Nor-Triterpenoids
Plants that are members of Schisandra genus are the source of propindilactone G and its derivatives, which constitute a family of polycyclic natural products [53][50]. Most members of this family are abundantly utilized in several medicines in China due to their high bioactive potency [54][51]. Propindilactone G along with its other derivatives are obtained from Schisandra propinqua var. propinqua, and they are structurally composed of fused cyclic rings. Owing to their frequent utilization in medicines, several research groups have devoted attention to their total synthesis. Wang et al. [55][52] in 2020 reported an efficient strategy for the total synthesis of propindilactone G. Total synthesis began with the Mitsunobu reaction of 68 followed by a reaction with N-bromosuccinimide and perchloric acid to prepare compound 69. It was then transformed into stable chlorohydrin, which was then joined with it to prepare compound 70 via a few steps. Compound 70 was then allowed to react with mesityl chloride in the presence of triethyl amine and tert-butanol followed by its treatment with 1,2 dimethoxyethane in the presence of DBU. It was then treated with Et3SiH, Co(thd)2 in the presence of dichloroethane and oxygen, which resulted in compound 71 in high yield (75%). Starting reagent 68 was reacted via several steps, thereby giving compound 72 in 78% yield. Compound 72 was then treated with p-TSA in the presence of toluene followed by Sharpless asymmetric dihydroxylation and treatment with HF and acetonitrile to obtain compound 73 in 96% yield. Compound 73 was converted to 74 via different steps, which then underwent Sharpless asymmetric dihydroxylation to give the target molecule, i.e., propindilactone G 75 in 58% yield (Scheme 11).
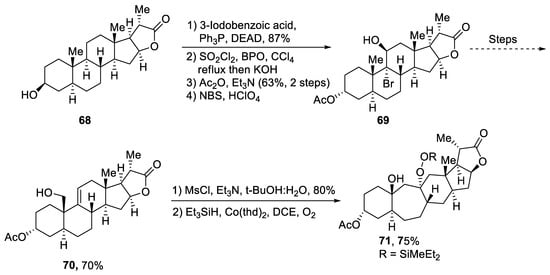
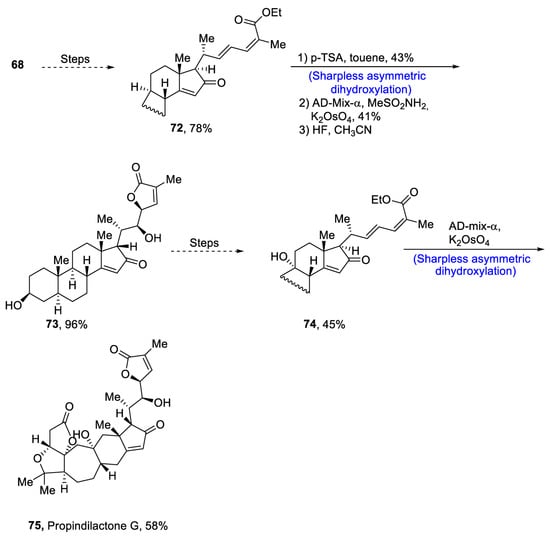
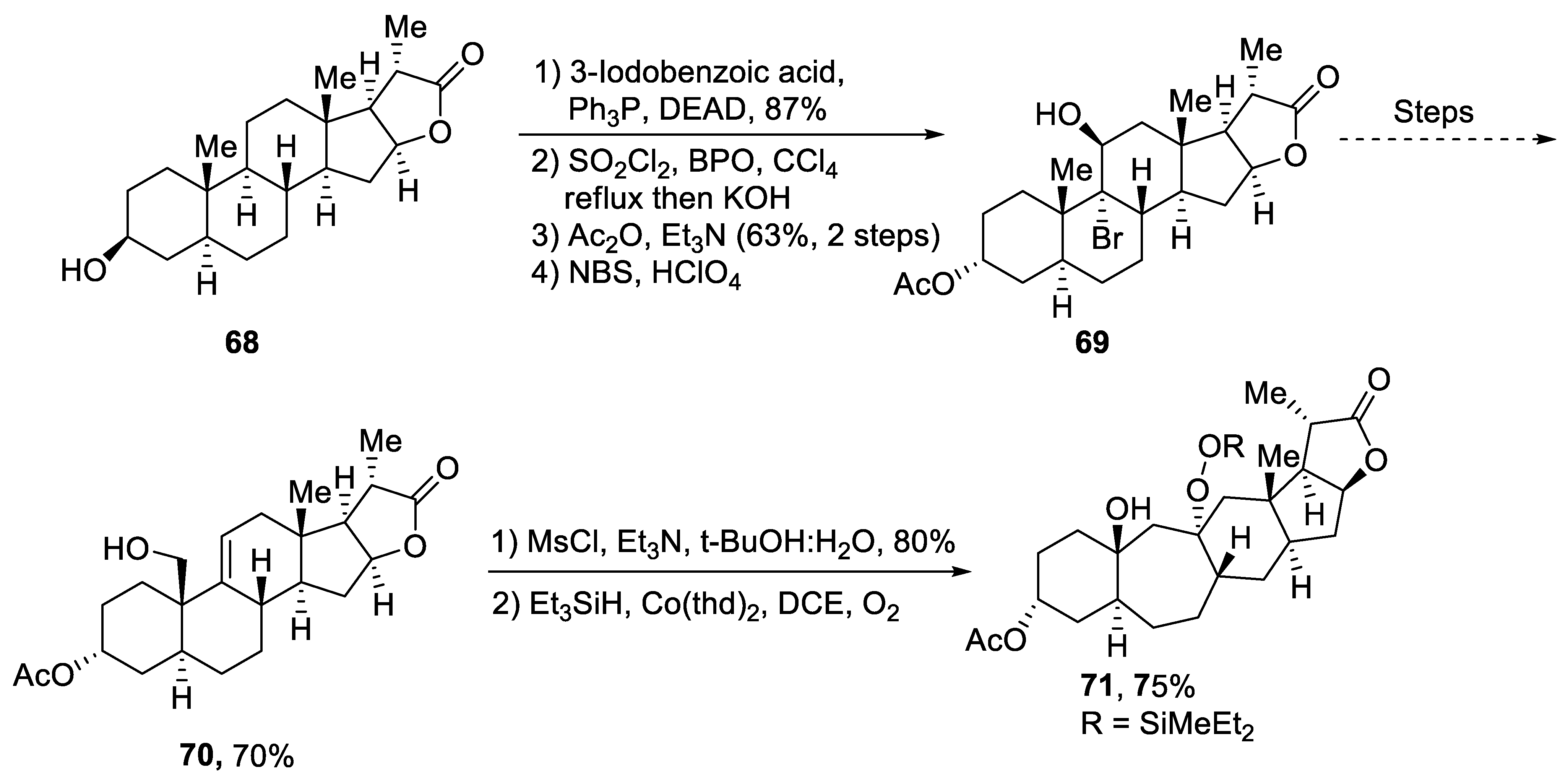
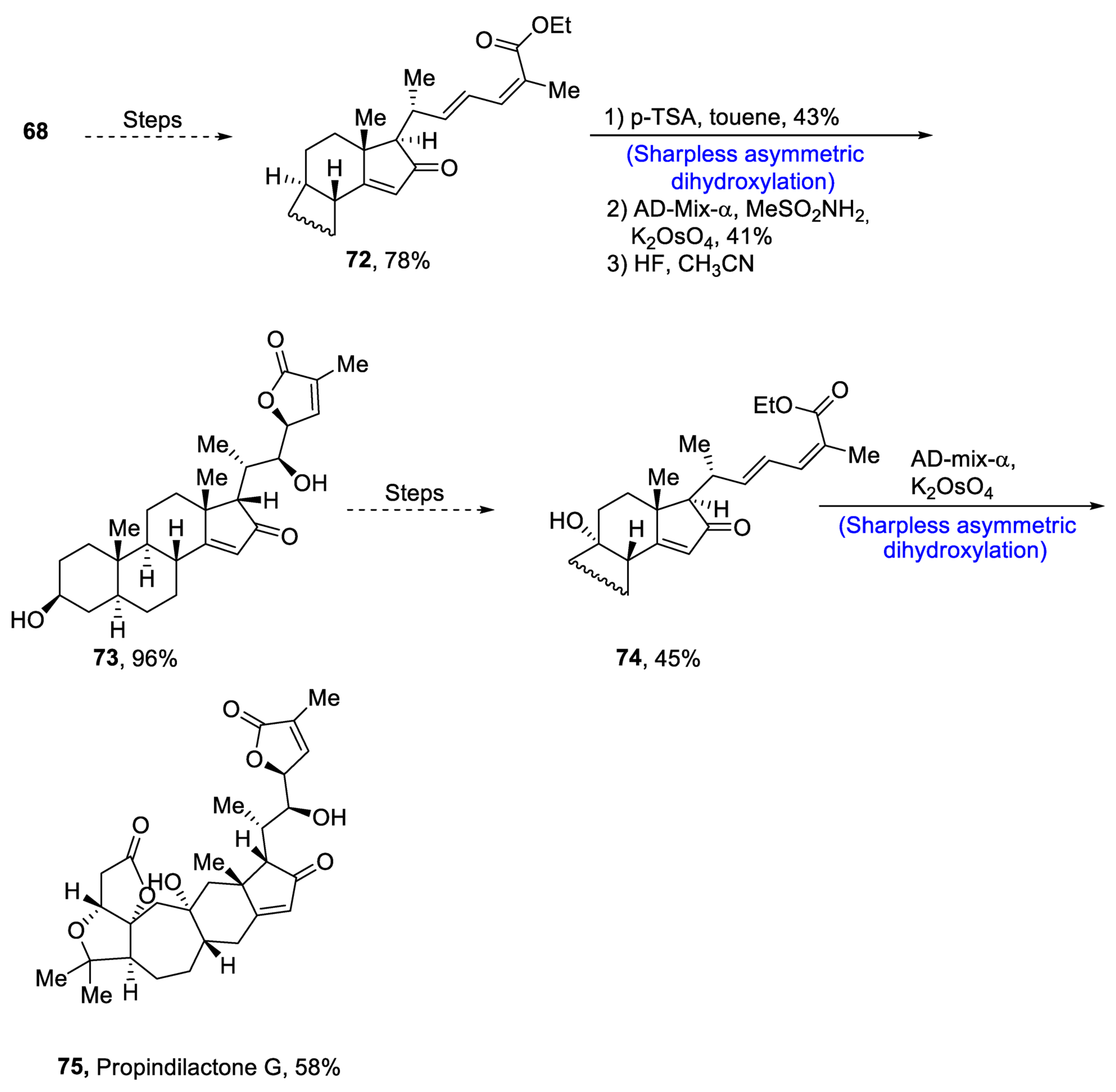
Scheme 11. Synthesis of propindilactone G 75.
3.4. Monoterpenoids
Aromatic 3,5-dimethylorsellinic acid (DMOA) is a source of many fungal monoterpenoids that constitute a variety of structures [56][53]. DMOA-isolated monoterpenoids, i.e., berkeyleyone and preaustinoids 1, preaustinoid 2 and preaustinoid 3, are biologically active and are highly potent against a number of inflammatory diseases [57][54]. (–)Berkeyleyone A structure is composed of a nonane core based on four cyclic rings. Keeping in view the medicinal importance of DMOA-derived (-)berkeyleyone A and preaustiniod 1-3, Zhang et al. [58][55] in 2021 described an efficient synthetic route towards their total synthesis. In this regard, 2,4,6-trihydroxybenzoic acid hydrate 76 was passed through four different steps and gave compound 77, which was subjected to reduction followed by treatment with 78 (which was obtained by Sharpless asymmetric dihydroxylation of 85) and later on, passing through sequential Krapcho dealkoxycarbonylation to obtain compound 79. Compound 79 was then subjected to Wittig olefination followed by base catalyzed acylation. Then, Krapcho-type demethylation was carried out followed by protection with trimethylsilyl group, which was then allowed to undergo Mander’s reagent-catalyzed acylation. Finally, deprotection of the silyl group led to the synthesis of (-)berkeleyone A 80 in 94% yield. Berkeyleyone A underwent Dess–Martin periodinane-mediated oxidation to synthesize preaustinoid A1 81 in 88% yield, which was then treated with boron trifluoride diethyl etherate in the presence of MeCN to synthesize preaustinoid B 83 in 86% yield, which was converted to preaustinoid B2 84 by the addition of aqueous NaOH and absolute alcohol. Similarly, preaustinoid A 81 was subjected to oxidation in the presence of m-chloroperoxy benzoic acid to yield preaustinoid A1 82 in 43% yield (Scheme 12).
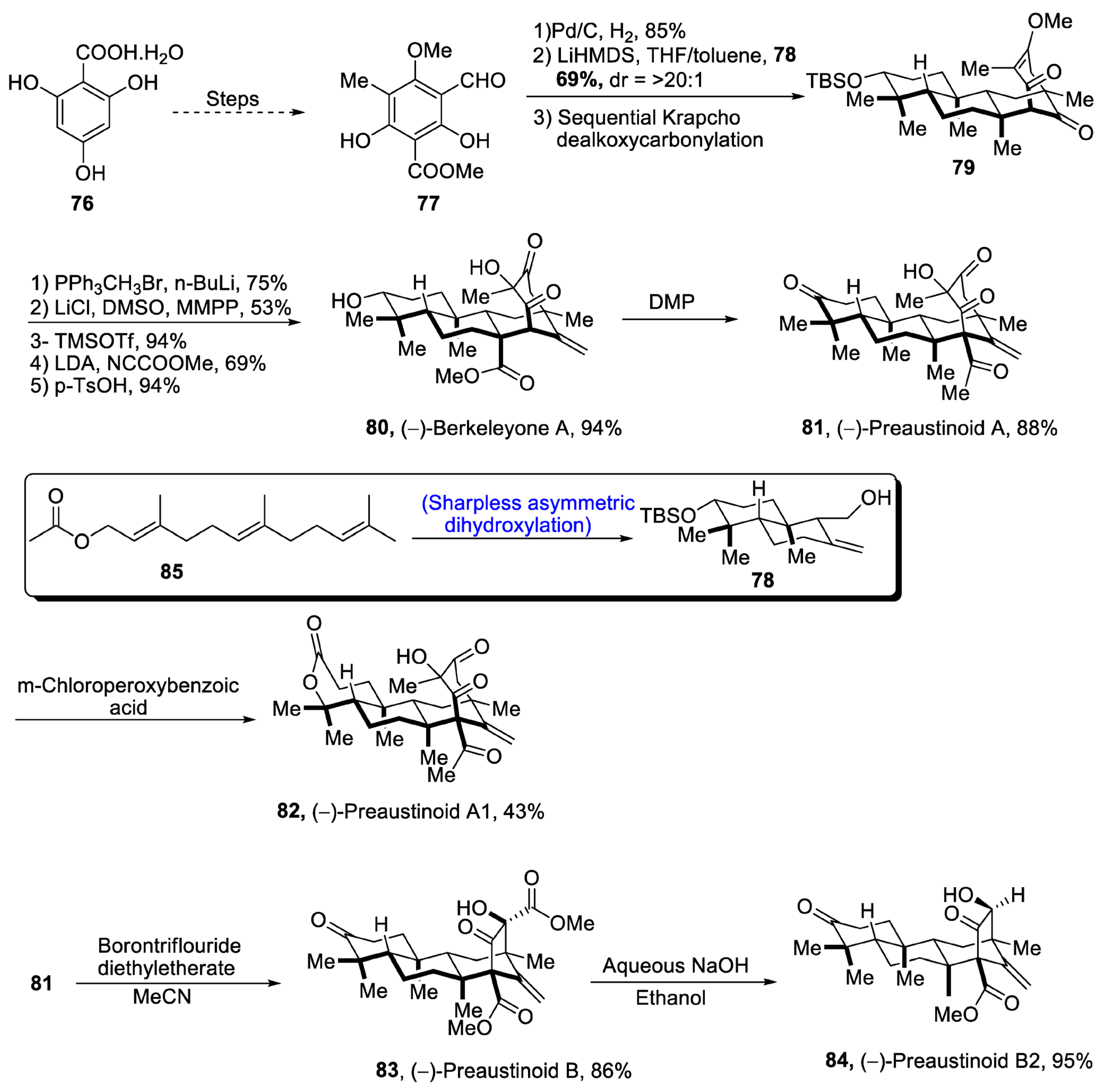
Scheme 12. Synthesis of berkeleyone A 80 and preaustinoids 81, 82, 83 and 84.
3.5. Monoterpenoid Alcohol
The new monoterpene alcohol was first isolated from a Chinese herb, namely ‘Mentha haplocalyx’, by Liu and his co-researchers [59][56]. However, ourthe recent studies do not correlate with their proposed structure, as it has been found to be incorrect via 13C and 1H-NMR spectroscopy. However, its structure was found to be quite similar to that of three other naturally occurring monoterpenes, namely asiasarinol, cosmosoxide B, and cis-p-menth-3-ene-1,2,8-triol [60][57]. This study [61][58] presented the total synthesis of trans-p-menth-3-ene-1,2,8-triol. The total synthesis began with the α and β-Sharpless asymmetric dihydroxylation, leading to the synthesis of compounds 87a and 87b in 40 and 43% yields with 33.8% and 1.2% enantiomeric excess, respectively, and similarly via route B, AD-mix-α and β leading to the synthesis of 90a and 90b in 76% and 91% yield with 54.5% ee and 59.4% enantiomeric excess, respectively. Compound 87a was further treated in the presence of methyl lithium and tetrahydrofuran followed by reaction with Dess–Martin periodinane and dichloromethane, thus finally converting to the synthesis of trans 88a (A-α) in 47% yield with 99.5% enantiomeric excess, by Luche reduction. On the other hand, other enantiomers of trans-monoterpenes were obtained by oxidation in the presence of manganese oxide followed by Luche reduction via both routes A and B. In this way, both enantiomers of the target molecule were obtained by using Sharpless asymmetric dihydroxylation (Scheme 13).
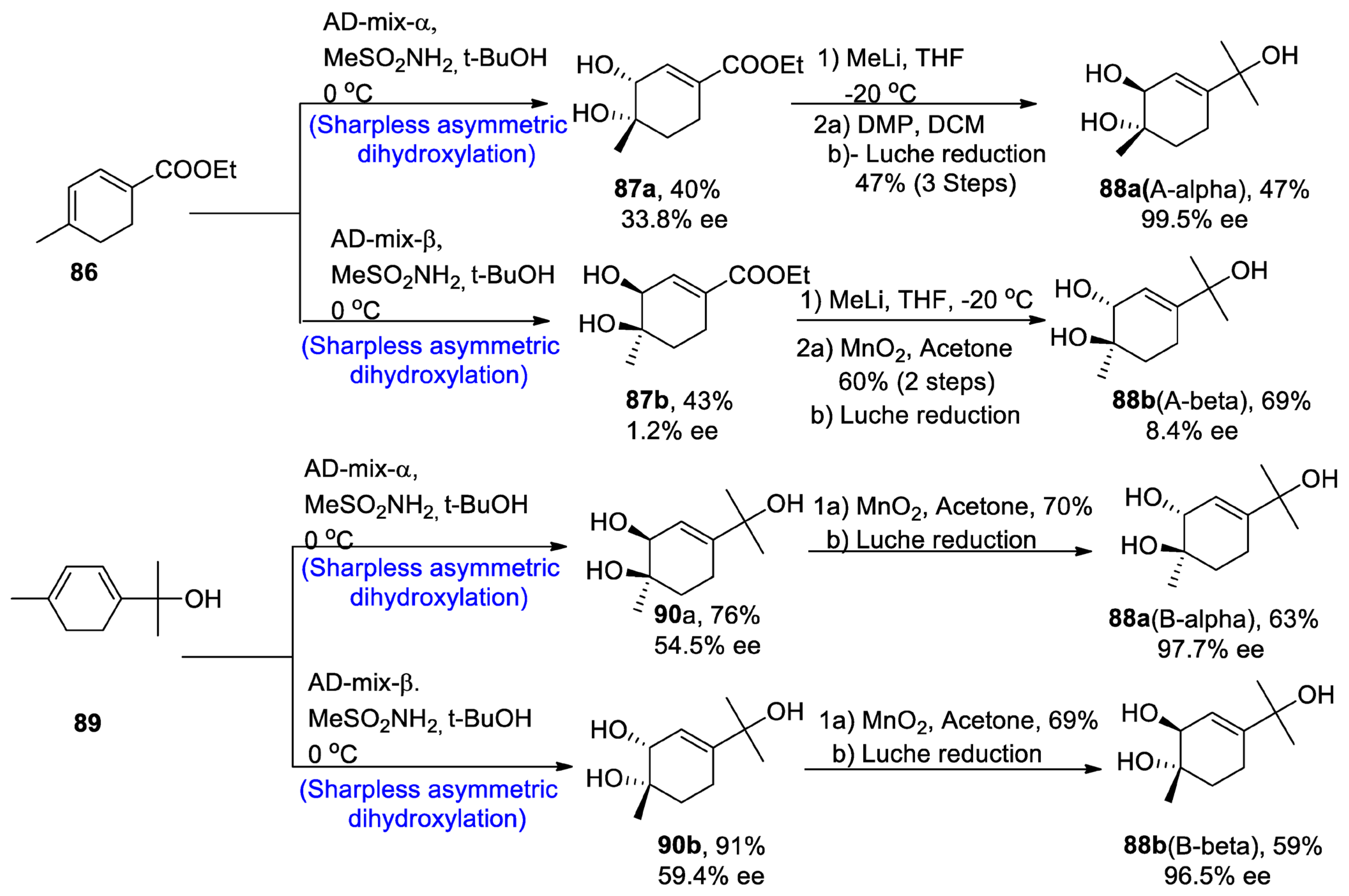
Scheme 13. Synthesis of trans-p-menth-3-ene-1,2,8-triol 88.
References
- Gnas, Y.; Glorius, F. Chiral auxiliaries—Principles and recent applications. Synthesis 2006, 12, 1899–1930.
- Kitano, Y.; Matsumoto, T.; Sato, F. A highly efficient kinetic resolution of γ- and β-trimethylsilyl secondary allylic alcohols by the sharpless asymmetric epoxidation. Tetrahedron 1988, 44, 4073–4086.
- Minato, M.; Yamamoto, K.; Tsuji, J. Osmium tetraoxide catalyzed vicinal hydroxylation of higher olefins by using hexacyanoferrate(III) ion as a cooxidant. J. Org. Chem. 1999, 55, 766–768.
- Martin, V.S.; Woodard, S.S.; Katsuki, T.; Yamada, Y.; Ikeda, M.; Sharpless, K.B. Kinetic resolution of racemic allylic alcohols by enantioselective epoxidation. A route to substances of absolute enantiomeric purity? J. Am. Chem. Soc. 1981, 103, 6237–6240.
- Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.S.; Kwong, H.L.; Morikawa, K.; Wang, Z.M. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. J. Org. Chem. 1992, 57, 2768–2771.
- Ogino, Y.; Chen, H.; Kwong, H.L.; Sharpless, K.B. On the timing of hydrolysis / reoxidation in the osmium-catalyzed asymmetric dihydroxylation of olefins using potassium ferricyanide as the reoxidant. Tetrahedron Lett. 1991, 32, 3965–3968.
- Hofmann, K.A. Sauerstoff-übertragung durch Osmiumtetroxyd und Aktivierung von Chlorat-Lösungen. Ber. Dtsch. Chem. Ges. 1912, 45, 3329–3336.
- Rheenen, V.V.; Kelly, R.C.; Cha, D.Y. An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Lett. 1976, 17, 1973–1976.
- Gao, Y. Osmium Tetroxide-t-Butyl Hydroperoxide. In Encyclopedia of Reagents for Organic Synthesis; John Wiley Sons, Ltd.: Hoboken, NJ, USA, 2001.
- Mehltretter, G.M.; Döbler, C.; Sundermeier, U.; Beller, M. An improved version of the Sharpless asymmetric dihydroxylation. Tetrahedron Lett. 2000, 41, 8083–8087.
- Hentges, S.G.; Sharpless, K.B. Asymmetric induction in the reaction of osmium tetroxide with olefins. J. Am. Chem. Soc. 1980, 102, 4263–4265.
- Sharpless, K.B.; Amberg, W.; Beller, M.; Chen, H.; Hartung, J.; Kawanami, Y.; Lubben, D.; Manoury, E.; Ogino, Y.; Shibata, T.; et al. New ligands double the scope of the catalytic asymmetric dihydroxylation of olefins. J. Org. Chem. 1991, 56, 4585–4588.
- Heravi, M.M.; Hajiabbasi, P. Recent advances in C-heteroatom bond forming by asymmetric Michael addition. Mol. Divers. 2014, 18, 411–439.
- Heravi, M.M.; Asadi, S.; Lashkariani, B.M. Recent progress in asymmetric Biginelli reaction. Mol. Divers. 2013, 17, 389–407.
- Heravi, M.M.; Zadsirjan, V. Recent advances in the application of the Oppolzer camphorsultam as a chiral auxiliary. Tetrahedron Asymmetry 2014, 25, 1061–1090.
- Heravi, M.M.; Hashemi, E. Recent applications of the Suzuki reaction in total synthesis. Tetrahedron 2012, 68, 9145–9178.
- Heravi, M.M.; Hashemi, E.; Azimian, F. Recent developments of the Stille reaction as a revolutionized method in total synthesis. Tetrahedron 2014, 70, 7–21.
- Heravi, M.M.; Hashemi, E.; Nzari, N. Negishi coupling: An easy progress for C–C bond construction in total synthesis. Mol. Divers. 2014, 18, 441–472.
- Heravi, M.M.; Hashemi, E.; Ghobadi, N. Development of Recent Total Syntheses Based on the Heck Reaction. Curr. Org. Syn. 2013, 17, 2192–2224.
- Heravi, M.M.; Lashaki, T.B.; Poorahmad, N. Applications of Sharpless asymmetric epoxidation in total synthesis. Tetrahedron Asymmetry 2015, 26, 405–495.
- Engler, E.M.; Andose, J.D.; Schleyer, V.R. Critical evaluation of molecular mechanics. J. Am. Chem. Soc. 1973, 95, 8005–8025.
- Blagg, B.S.J.; Boger, D.L. Total synthesis of (+)-camptothecin. Tetrahedron 2002, 58, 6343–6349.
- Ghosal, S.; Saini, K.S.; Razdan, S. Crinum alkaloids: Their chemistry and biology. Phytochemistry 1985, 24, 2141–2156.
- Pettit, G.; Orr, B.; Ducki, S. Anticancer, pancratistatin, phosphate prodrugs, phosphorylation. Anti-Cancer Drug Design 2000, 15, 389–395. Available online: https://www.ingentaconnect.com/contentone/cog/antcan/2000/00000015/00000006/art00001 (accessed on 12 March 2022).
- Zhao, Y.; Zhu, Y.; Ma, G.; Wei, Q.; Yang, S.; Zeng, X.; Zhang, H.; Chen, J. Short, Enantioselective, gram-scale synthesis of (−)-zephyranthine. Chem. Sci. 2021, 12, 9452–9457.
- Zhou, Z.F.; Menna, M.; Cai, Y.S.; Guo, Y.W. Polyacetylenes of Marine Origin: Chemistry and Bioactivity. Chem. Rev. 2015, 115, 1543–1596.
- Hartung, J.; Kneuer, R. Synthesis of enantiopure (2R)-configured muscarine alkaloids via selective alkoxyl radical ring-closure reactions. Tetrahedron Asymmetry 2003, 14, 3019–3031.
- Gehlawat, A.; Prakash, R.; Pandey, S.K. A Short and Efficient Enantioselective Synthesis of (+)-(2S,3S,5S)-epi-Muscarine. Chem. Sel. 2020, 5, 6373–6375.
- Zhang, L.; Zhang, C.J.; Zhang, D.B.; Wen, J.; Zhao, X.W.; Li, Y.; Gao, K. An unusual indole alkaloid with anti-adenovirus and anti-HSV activities from Alstonia scholaris. Tetrahedron Lett. 2014, 55, 1815–1817.
- Cai, Y.S.; Sarotti, A.M.; Zhou, T.L.; Huang, R.; Qiu, G.F.; Tian, C.K.; Miao, Z.H.; Mandi, A.; Kurtan, T.; Cao, S.G.; et al. Flabellipparicine, a flabelliformide-apparicine-type bisindol alkaloid from Tabernaemontana divaricate. J. Nat. Prod. 2018, 81, 1976–1983.
- Pourroy, B.; Honore, S.; Pasquier, E.; Rey, B.V.; Kruczynski, A.; Briand, C.; Braguer, D. Antiangiogenic concentrations of vinflunine increase the interphase microtubule dynamics and decrease the motility of endothelial cells. Cancer Res. 2006, 66, 3256–3263.
- Zhang, J.; Song, M.; Ao, Y.L.; Li, Y.; Zou, X.Y.; Xu, J.; Wang, Y.; Zhang, D.M.; Zhang, X.Q.; Ye, W.C. Alstolarines A and B, Two Unusual Monoterpenoid Indole Alkaloids with Acetal Moiety from Alstonia scholaris. Org. Chem. Front. 2020, 7, 3468–3473.
- Gossan, D.P.A.; Alabdul Magid, A.; Yao, P.A.K.; Behr, J.B.; Ahibo, A.C.; Djakouré, L.A.; Harakat, D.; Nazabadioko, L.V. Glycosidase inhibitors from the roots of Glyphaea brevis. Phytochemistry 2015, 109, 76–83.
- Shibano, M.; Tsukamoto, D.; Kusano, G. Polyhydroxylated Alkaloids with Lipophilic Moieties as Glycosidase Inhibitors from Higher Plants. Heterocycles 2002, 57, 1539–1553. Available online: https://cir.nii.ac.jp/crid/1521417755858432768 (accessed on 12 March 2022).
- Byatt, B.J.; Kato, A.; Pyne, S.G. Synthesis and Structural Revision of Glyphaeaside C. Org. Lett. 2021, 23, 4029–4033.
- Ratnayake, R.; Covell, D.; Ransom, T.T.; Gustafson, K.R.; Beutler, J.A. Englerin A, a Selective Inhibitor of Renal Cancer Cell Growth, from Phyllanthus engleri. Org. Lett. 2009, 11, 57–60.
- Sourbier, C.; Scroggins, B.T.; Ratnayake, R.; Prince, T.L.; Lee, S.; Lee, M.J.; Nagy, P.L.; Lee, Y.H.; Trepel, J.B.; Beutler, J.A.; et al. Englerin A Stimulates PKCθ to Inhibit Insulin Signaling and to Simultaneously Activate HSF1: Pharmacologically Induced Synthetic Lethality. Cancer Cell 2013, 23, 228–237.
- Mou, S.B.; Xiao, W.; Wang, H.Q.; Wang, S.J.; Xiang, Z. Syntheses of Epoxyguaiane Sesquiterpenes (−)-Englerin A, (−)-Oxyphyllol, (+)-Orientalol E, and (+)-Orientalol F: A Synthetic Biology Approach. Org. Lett. 2020, 22, 1976–1979.
- Vichnewski, W.; Sarti, S.J.; Gilbert, B.; Herz, W. Goyazensolide, a Schistosomicidal Heliangolide from Eremanthus-Goyazensis. Phytochemistry 1976, 15, 191–193.
- Acuna, U.M.; Shen, Q.; Ren, Y.; Lantvit, D.D.; Wittwer, J.A.; Kinghorn, A.D.; Swanson, S.M.; Carcach de Blanco, E.J. Goyazensolide Induces Apoptosis in Cancer Cells in vitro and in vivo. Int. J. Cancer Res. 2013, 9, 36–53. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4303185/ (accessed on 12 March 2022).
- Liu, W.; Patouret, R.; Barleunga, S.; Plank, M.; Loewith, R.; Winssinger, N. Identification of a Covalent Importin-5 Inhibitor, Goyazensolide, from a Collective Synthesis of Furanoheliangolides. ACS Cent. Sci. 2021, 7, 954–962.
- Maurer, B.; Grieder, A. Sesquiterpenoids from Costus Root Oil (Saussurea lappa CLARKE). Helvetica 1977, 60, 2177–2190.
- Seiichi, T.; Takumichi, S.; Kiyohiro, S.; Masashi, A.; Kunio, O. Enantiodivergent Route to the Aromatic Bisabolane Sesquiterpenes via a Chiral Acetylene Alcohol. Chem. Lett. 1989, 18, 1781–1784.
- Yajima, A.; Shirakawa, I.; Shiotani, N.; Ueda, K.; Murakawa, H.; Saito, T.; Katsuta, R.; Ishigami, K. Practical synthesis of aromatic bisabolanes: Synthesis of 1,3,5-bisabolatrien-7-ol, peniciaculin A and B, and hydroxysydonic acid. Tetrahedron 2021, 92, 132253.
- Ushakov, D.B.; Navickas, V.; Strobele, M.; Mossmer, C.M.; Sasse, F.; Maier, M.E. Total Synthesis and Biological Evaluation of (−)-9-Deoxy-englerin A. Org. Lett. 2011, 13, 2090–2093.
- Peng, G.P.; Tian, G.; Huang, X.F.; Lou, F.C. Guaiane-type sesquiterpenoids from Alisma orientalis. Phytochemistry 2003, 63, 877–881.
- Palli, K.K.; Anugu, R.R.; Chandrasekhar, S. Total Synthesis of (−)-4- epi -Englerin A. Eur. J. Chem. 2021, 22, 31990–33196.
- Alvarado, R.B.H.; Mazon, A.M.; Ortega, A.; Mayorga, K.M. DARK Classics in Chemical Neuroscience: Salvinorin A. ACS Chem. Neurosci. 2020, 11, 3979–3992.
- Zimdars, P.; Wang, Y.; Metz, P. A Protecting-Group-Free Synthesis of (-)Salvinorin A. Chem. A Eur. J. 2021, 27, 7968–7973.
- Xiao, W.L.; Li, R.T.; Huang, S.X.; Pu, J.X.; Sun, H.D. Triterpenoids from the Schisandraceae family. Nat. Prod. Rep. 2008, 25, 871–891.
- Li, X.; Cheong, P.H.Y.; Carter, R.G. Schinortriterpenoids: A case study in synthetic design. Angew. Chem. Int. Ed. 2017, 56, 1704–1718.
- Wang, Y.; Chen, B.; He, X.; Gui, J. Bioinspired Synthesis of Nortriterpenoid Propindilactone G. J. Am. Chem. Soc. 2020, 142, 5007–5012.
- Santos, R.M.G.D.; Fo, S.R. Meroterpenes from Penicillium sp found in association with Melia azedarach. Phytochemistry 2002, 61, 907–912.
- Guo, C.J.; Knox, B.P.; Chiang, Y.M.; Lo, H.C.; Sanchez, J.F.; Lee, K.H.; Okaley, B.R.; Bruno, K.S.; Wang, C.C.C. Molecular Genetic Characterization of a Cluster in A. terreus for Biosynthesis of the Meroterpenoid Terretonin. Org. Lett. 2012, 14, 5684–5687.
- Zhang, Y.; Ji, Y.; Franzoni, I.; Guo, C.; Jia, H.; Hong, B.; Li, H. Enantioselective Total Synthesis of Berkeleyone A and Preaustinoids. Angew. Chem. Int. Ed. 2021, 60, 14869–14874.
- She, G.; Xu, C.; Liu, B.; Shi, R. Two new monoterpenes from Mentha haplocalyx Briq. Helv. Chim. Acta 2010, 93, 2495–2498.
- Konishi, S.; Mori, N.; Takikawa, H. Synthesis guided structure revision of the monoterpene alcohol isolated from Mentha haplocalyx. Biosci. Biotechnol. Biochem. 2019, 83, 391–399.
- Konishi, S.; Ogura, Y.; Takikawa, H.; Watanabe, H. Asymmetric synthesis of trans-p-menth-3-ene-1,2,8-triol, the monoterpene isolated from herbal plants. Biosci. Biotechnol. Biochem. 2020, 84, 37–42.
More