In the context of the growing human demand for green secondary energy sources, proton-exchange membrane water electrolysis (PEMWE) is necessary to meet the high-efficiency production of high-purity hydrogen required for proton-exchange membrane fuel cells (PEMFCs). The development of stable, efficient, and low-cost oxygen evolution reaction (OER) catalysts is key to promoting the large-scale application of hydrogen production by PEMWE.
1. Introduction
Due to the non-renewable nature of fossil fuels, excessive consumption of fossil fuels for energy is bound to cause serious ecological problems
[1]. According to a report by the International Energy Agency (IEA), total global energy demand will exceed the equivalent of 18 billion tonnes of oil by 2030
[2]. Therefore, the development of secondary energy is crucial to meet the needs of sustainable development. The need for efficient production of renewable, non-polluting secondary energy is also urgent to meet people’s quest for a higher standard of living while preserving the ecological environment. Recently, renewable energy systems such as solar power, wind and tidal energy are considered as potential green secondary energy. However, the large-scale application of these energy conversion devices has been hindered by the limitations of their geographical nature, periodicity and high cost of long-distance transmission
[3][4][5][3,4,5].
As a power generation device, proton-exchange membrane fuel cells (PEMFCs) have attracted various attention. PEMFCs can efficiently convert the chemical energy of hydrogen and oxygen directly into electrical energy
[6]. On the other hand, PEMFCs have lower energy losses and higher energy conversion efficiencies because they do not need to go through the Carnot cycle to convert energy
[7]. On the other hand, PEMFCs also have the advantage of zero missions, as the only running product is pure water. The raw material required for the PEMFCs is high-purity hydrogen. Oetjen et al. found that for Pt-based catalysts in PEMFCs, even if exposed to feed gas containing 100 ppm CO for 5 min, the catalytic performance would significantly decline
[8]. Currently, conventional hydrogen production processes are still based on natural gas reforming. However, hydrogen obtained through the natural gas reforming process could not meet the standard required for PEMFCs application, due to its poor purity
[2][9][2,9]. It would also be impractical to purify the hydrogen, which would add to the cost of production. The purity of hydrogen produced by proton-exchange membrane water electrolysis (PEMWE) is up to 99.99%, which has obvious advantages over other hydrogen production methods
[10]. Therefore, for PEMFCs to be deployed on a large scale, the promotion of PEMWE is of great importance.
The hydrogen produced by PEMWE is an ideal source of feed gas for the large-scale production of PEMFCs. PEMWE has the advantages of compact design, small footprint, high current density
[11][12][11,12]. In practice, noble metals like Ir and Ru catalysts are heavily used in the OER catalysts designed to lower the potential barrier, leading to the high cost of PEMWE. According to the report, the cost of hydrogen production with PEMWE is as high as ~$10.30/kg, and only ~4% of the hydrogen is produced through PEMWE, due to the high cost
[13]. Therefore, it is of great significance to design cost-effective, efficient and highly stable OER catalysts.
2. Fundamentals and Current Status of Research on Acidic OER
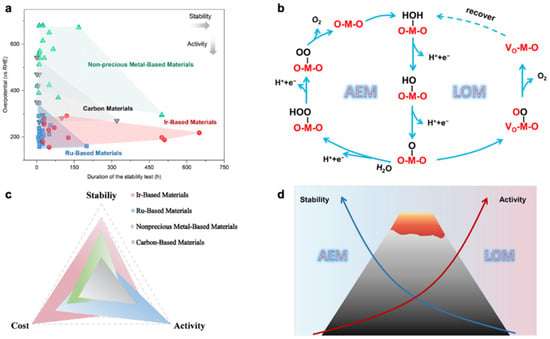
Although the O
2 produced by the OER reaction is a by-product of the electrolysis of water for hydrogen production, the reaction kinetics are slower in an acidic environment due to the fact that OER is a four-electron process and requires higher energy to overcome the high potential barrier of the kinetics
[14][21]. As a result, the anode reaction consumes the most energy process in the entire electrolytic water process. In order to reduce the overpotential, large amounts of precious metal are used in the anode catalysts. Ir-based and Ru-based catalysts exhibit impressive catalytic activity for OER reactions in acidic media
[15][22]. It is now generally accepted that the acidic OER process is dominated by two mechanistic routes, the adsorbed evolution mechanism (AEM) and lattice oxygen mechanism (LOM)
[16][23]. As shown in
Figure 1b, the OER process through AEM involves four steps and proceeds on a single adsorption site. There is a linear relationship between the adsorption energies of O*, *OH and *OOH
[17][24]. Utilizing isotopic labelling, researchers have investigated the contribution of lattice oxygen to OER. The results show that the acidic OER process is co-dominated by both AEM and LOM. The proportion of LOM is related to the catalyst structure
[18][19][25,26]. The LOM process typically has a lower activation barrier than the AEM process. However, the LOM process can lead to degradation of the active component of the catalyst, resulting in reduced stability of the OER catalyst
[16][20][23,27].
Figure 1. (
a) Stability test time and ŋ10 of catalysts reported in recent years and (
b) acid OER reaction mechanism. (
c) The stability, activity, and cost of Ir-, Ru-, nonprecious metal-, and carbon-based materials. (
d) Schematic diagram of the difference in stability and activity of the AEM/LOM mechanism
[17][24].
Ir and Ru are noble metals, which is the main cost of electrolytic cells. This has become the biggest obstacle to the industrialization of PEMWE. However, in acidic media and under high-voltage conditions, both Ru-based and Ir-based catalysts are not sufficiently stable
[21][22][23][28,29,30], which is not sufficient for the large-scale application of PEMWE. Great efforts have been made to improve the atomic utilization of precious metals and to enhance the performance and stability of acidic OER catalysts. Ru/Mo
2C heterojunctions were constructed to regulate the valence of Ru and Mo in the catalyst to simultaneously boost OER and HER in acidic conditions
[24][31]. A chemical etching strategy for fabricating a Ru/Fe oxide towards OER resulted in enhanced stability
[25][32]. Sr–Ru–Ir ternary oxide electrocatalysts: The electronic structure of active Ru sites was modulated by Sr and Ir, resulting in an overpotential of 190 mV at 10 mA cm
−2, and the overpotential remained below 225 mV following 1500 h of operation
[23][30]. Rh-Ir alloy NPs prepared by a scalable microwave-assisted method with improved atom utilisation through ~10 nm ultra-fine size: Owing to the synergy of ensemble and electronic effects by alloying a small amount of Rh with Ir, the binding energy difference of the O and OOH intermediates was reduced, leading to faster kinetics and enhanced OER activity
[26][33].
It has also been shown that alloys
[27][28][29][34,35,36], perovskite material
[30][31][32][33][37,38,39,40], single-atomic materials
[34][35][36][37][41,42,43,44], MOF materials
[38][39][40][41][45,46,47,48], and transition metal oxides
[42][43][49,50] have potential applications in acidic OER. Unfortunately, as is shown in
Figure 1c, due to the harsh acidic conditions, it seems difficult to accomplish the balance of stability and catalytic performance with these reported non-precious metal catalysts. Therefore, the current position of noble metal catalysts in OER catalysts is still unshakable. Due to the high cost of noble metals, improving catalytic activity and durability of catalysts while reducing the amount of noble metals is the focus of current research. To further improve the industrial viability of PEMWE, researchers have explored many strategies that can significantly enhance the catalytic activity and stability of Ru-based and Ir-based catalysts for OER in acidic media, such as doping
[44][45][46][51,52,53], morphological engineering
[47][48][54,55], defect engineering
[49][50][56,57], etc.
3. Support-Catalyst Interactions in Acidic OER
3.1. Metal Support Interaction (MSI)
MSI is commonly found in supported metal-based catalysts. Metal NPs are a fundamental component of various catalysts due to their unique electronic structure, morphology and controlled composition. Immobilization of metal NPs on support could improve their stability and control their spatial distribution. In addition, since carriers are usually not inert, the interaction between carriers and metal NPs can generate specific interfacial phenomena
[51][62].
3.1.1. Charge Transfer
Due to the difference in Fermi energy levels between metal NPs and supports, interfacial contact between NPs and supports can induce changes in the electronic structure of both supports and NPs to seek an equilibrium in the electrochemical potential, and such changes are bi-directional in nature
[52][63]. The metallic properties of the NPs give the electrons mobility. However, the mobility is related to the nanosystem because the smaller the NP, the more localized its electronic state is. In some cases, the charge transfer may be accompanied by a change in the oxidation state of the NPs or the metal ions of the supports
[53][64].
3.1.2. Interfacial Perimeter
The interfacial region around the NPs forms a unique environment as the NPs, support and reactants make direct contact and synchronously promote the catalytic reaction. As a result, it can significantly enhance the adsorption and reactions of molecules at the perimeter
[54][66]. The close proximity of NPs to different groups or defects (e.g., oxygen vacancies, hydroxyl groups, Lewis acids or Lewis bases) on the surface of the support may also contribute to localized sequential reactions of reactants or products, or the stabilization of transition states
[38][45]. While this phenomenon of MSI can modulate the intensity of oxygenated intermediates adsorption by the catalyst, it accelerates the kinetics of the catalyst’s reaction to the OER process.
3.1.3. Nanoparticle Morphology
The shape and crystal structure of NPs have a strong influence on their catalytic performance. This is because NPs of different morphologies expose distinct crystalline facets, which can result in either favorable or unfavorable atomic configurations in different reactions. Therefore, the activity and selectivity of catalysts can be effectively modulated by modulating the morphology of NPs at the metal-support interface
[55][68]. Supported iridium- and ruthenium-based NPs are potential OER catalysts for PEMWE, but the inhomogeneous dispersion of these NPs on the support and their inhomogeneous size usually leads to migration and adhesion of the catalyst at high potentials and strong acidity. Ultimately, this leads to a loss of active surface area and catalytic material, resulting in reduced OER performance.
3.1.4. Chemical Composition
Solid-state reactions can occur between the metal NPs and the support, resulting in the formation of a new phase. The exchange of species is possible in both directions and is usually designed for redox reactions
[55][68]. The local composition of alloyed metal NPs can be affected by interactions with the support, where the rearrangement of components in alloyed NPs is driven by the interaction of their elements with the support at the interface. This can lead to compositional rearrangements that differ from the initial homogeneous composition, such as sub-nanostructures like core-shells, affecting the synergy between the metal and metal active sites and thus, their catalytic performance (
Figure 2)
[56][70].
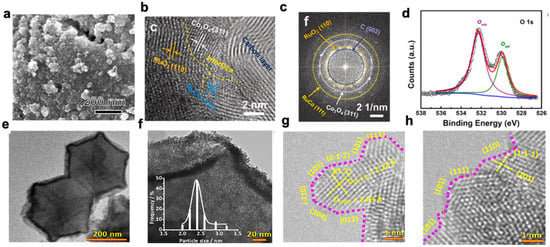
Figure 2. (
a) SEM images of RuCoO
x-RuCo-NC; (
b) line profiles of RuCoO
x-RuCo-NC; (
c) HAADF-STEM elemental mappings; (
d) XPS spectra of RuCoO
x-RuCo-NC: O 1s
[54][66]; (
e) SEM image of RuIr@CoNC catalysts; (
f) size distribution of RuIr nanocrystals; (
g,h) HRTEM images. Atomic steps are indicated with pink dotted lines
[57][69].
3.2. Strong Metal-Support Interaction (SMSI)
Although SMSI is also an MSI phenomenon, because it has special characteristics compared to the other four common MSI phenomena, it will be discussed separately. SMSI was first proposed by Tauster et al. in 1978
[56][70]. Since then, researchers have conducted in-depth studies on the formation mechanism of SMSI, and it is generally agreed that one of the main reasons for the formation of SMSI is the different surface energy and work functions between metal NPs and supports, which drive the surface migration of the supports to the metal NPs, thus stabilizing the metal NPs
[55][68]. SMSIs are normally differentiated according to the different construction methods. SMSIs constructed using high temperature reduction are referred to as classical SMSIs
[56][70]; those constructed with an oxygen atmosphere are referred to as O-SMSIs
[58][71]; and those formed using adsorbent induction are referred to as A-SMSIs
[59][72]. Although there are various methods of SMSI construction, there is still a lack of effective methods to purposefully construct SMSI on some relatively inert oxide supports such as SiO
2, CaO, Al
2O
3 and so on
[60][73]. The use of SMSI can effectively modulate the distribution, valence, and ligand structure and stability of NPs on the surface of the support, creating great opportunities for the electronic structure modulation of the active component of the catalyst
[60][73]. Xu et al. synthesized a series of M-Ru/RuO
2@CNT (M = Mn, Cd, Cu) bifunctional OER/HER catalysts. The OER activity and stability of the catalysts were improved by strong SMSI. The optimized Mn-Ru/RuO
2@CNT catalyst with an ultra-small particle size of 2.5 nm showed the best catalytic performance, with a low overpotential of 177 mV and 30 mV for OER and HER, respectively, at a current density of 10 mA cm
−2 in 0.5 M H
2SO
4 [61][74].
(1) The migration of the support to the surface of the metal particles to form an inclusion structure. Zhang et al. constructed a classical SMSI by high temperature treatment of Au/TiO
2. The presence of an amorphous inclusion layer was observed by HETEM and the chemical components of the inclusion layer were analyzed by EELS
[62][75]. The inclusion layer acted as immobilization and protection for the NPs. In the A-SMSI constructed in Rh/TiO
2, the Rh NPs were wrapped by amorphous TiO
x and the inclusions were composed of Ti species of different valence states (Ti
3+/Ti
4+=3/7), whereas in the Rh/TiO
2 catalyst with classical SMSI effect, the Rh NPs were wrapped by a more crystalline TiO
x diatomic layer and only Ti
3+ were present in the inclusions
[59][72].
(2) Intermetallic bonding. Tauster et al. suggested that intermetallic bonding was facilitated by the overlap of the d and empty d orbitals of the excess metal between the active component and the support
[56][70]. An Au/ZnO catalyst reported by Chung et al. showed that Au-Zn interactions could be formed by treating Au/ZnO with oxygen at different temperatures by fine structure resolution of Au by EXAFS. The formation of Au-Zn bonds (similar to AuZn alloys) can be directly detected after reduction at 300 °C under hydrogen atmosphere
[58][71]. The phenomenon of intermetallic bonding caused by SMSI can be a rational strategy for researchers to regulate the electronic structure of active components in OER catalysts. In response to the limitations of the conventional AEM and LOM evolutionary mechanisms on the performance as well as the stability of acidic OER catalysts, Lin et al. innovatively reported an OER electrocatalyst (Ru/MnO
2) with Ru atomic array patches supported on α-MnO
2. The OER catalytic mechanism of this catalyst involves only *O and *OH species as intermediates. This mechanism allows the direct coupling of O-O radicals to generate and precipitate O
2. Ru/MnO
2 exhibits high OER activity (161 mV@10 mA cm
−2) and excellent stability (minimal loss of activity after 200 h of operation) and is one of the best-performing catalysts available for acid-stable oxygen precipitation reactions.
(3) Modification of the adsorption characteristics of the catalytical active component. As early as 1982, Burch et al. found that the adsorption of H
2 by the catalyst material was substantially reduced after high temperature reduction of Ni/TiO
2 at 650 °C
[63][78]. It has also been mentioned in the literature that O-SMSI and A-SMSI constructed by oxygen treatment and adsorbent induction lead to a significant reduction in the adsorption capacity of the material for small molecules such as CO and H
2 [59][64][72,79]. The property that SMSI can change the adsorption characteristics of the material allows one to design SMSI to adjust the adsorption energy of the catalyst material for OER reactions on intermediates, thus improving the catalytic effect of the catalyst and even improving the stability of the catalyst. According to the OER volcanogram, tungsten oxide has a relatively weaker O-binding capacity than iridium oxide, and it is thus possible to coordinate the O-binding capacity by constructing a W-Ir dual active site on the catalyst
[65][80]. However, the actual catalytic effect of tungsten oxide in acidic OER is not satisfactory, due to its degradation by intermediates in the OER reaction
[66][81].
3.3. Strong Oxide-Support Interaction (SOSI)
Initially, the SMSI was primarily concerned with strong interactions between metal atoms and supports, but this was gradually extended to strong interactions between metal-containing species and supports
[67][84]. The concept of SOSI was derived to better describe the strong interactions between oxides and supports in order to distinguish them from SMSI. In general, similar to those of SMSI, SOSI has the following effects.
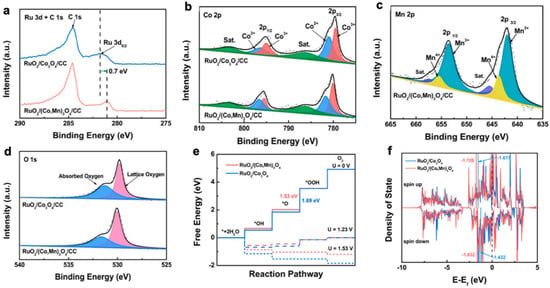
(1) SOSI can redistribute electrons at the oxide-support interface and modulate the electronic properties of the catalytic active site. Niu et al. reported the synthesis of a RuO
2/(Co, Mn)
3O
4 nanocomposite. The introduction of manganese in Co
3O
4 resulted in the redistribution of support electrons. By XPS characterization, the
scautho
lars found that RuO
2/(Co, Mn)
3O
4 catalysts appeared as electron-rich Ru species. The adsorption of O on RuO
2/(Co, Mn)
3O
4 was weakened as the electronic properties changed, which resulted in the rate-determining step in the OER process, i.e., the formation of OOH*, being accelerated. This cost-effective catalyst (ultra-low Ru content of 2.51 wt%) requires an overpotential of only 270 mV to achieve a current density of 10 mA cm
−2 with a mass activity nearly 69 times better than that of commercial RuO
2 (
Figure 34)
[68][85].
Figure 34. XPS spectra of (
a) Ru 3d, (
b) Co 2p, (
c) Mn 2p, and (
d) O 1s of RuO
2/Co
3O
4/CC and RuO
2/(Co, Mn)
3O
4/CC samples. (
e) Gibbs free energy diagrams of supported catalysts. (
f) Projected density of states (PDOS) of Ru d orbitals in supported catalysts
[68][85].
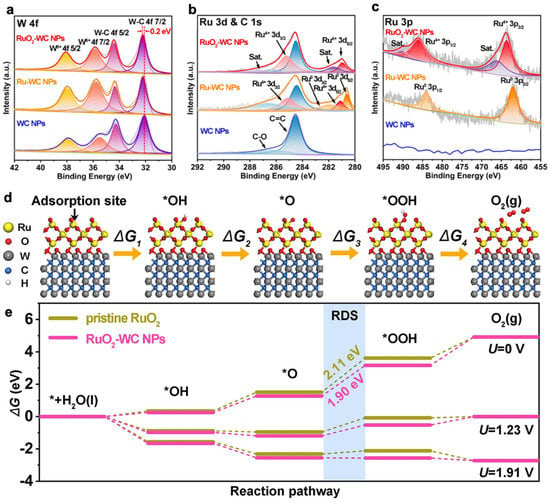
(2) SOSI can also effectively reduce the aggregation of NPs in electrocatalytic reactions, thereby improving the atomic utilization and stability of the material. WC-loaded RuO
2 NPs (RuO
2-WC NPs) anchored on carbon nanosheets reported by Sun et al. exhibited strong catalyst-support interactions and the low loading of Ru (4.11 wt.%) significantly enhanced the acidic oxygen precipitation reaction activity. The 10 mA/cm
2 overpotential was 347 mV and the mass activity was eight times higher than that of commercial RuO
2. The excellent OER performance can be attributed to the catalyst-support interaction between RuO
2 and the WC support. Theoretical calculations show that the strong catalyst-support interaction between RuO
2 and WC supports optimizes the electronic structure around the Ru site, giving good adsorption energy for OER intermediates and thus reducing the reaction potential of the RDS.
(3) SOSI can improve the catalytic performance of OER catalytic materials by building rich interfaces, thus improving the catalytic performance of OER catalytic materials. Highly dispersed IrO
2 nanoclusters (~1 nm) loaded on porous V
2O
5 supports were successfully prepared by Zheng et al. using a MIL-88B(V) organometallic framework as a self-sacrificing template. Through detailed characterization and comparative experiments, the strong interaction between IrO
2 nanoclusters and V
2O
5 supports resulted in significant lattice distortion of IrO
2 nanoclusters, which facilitated the exposure of more unsaturated coordination active sites. At the same time, the strong electron transfer (electron transfer from Ir to V) at the interface between IrO
2 and V
2O
5 allows the IrO
2 active sites to act as electrophilic centers, weakening the adsorption of oxygen-containing intermediates. DFT calculations suggested that the SOSI effect alters the occupation number of Ir eg orbitals, lowering the *O to *OOH energy barrier and thus effectively promoting the reaction rate of water oxidation. As a result, the IrO
2/V
2O
5 catalyst exhibited excellent OER activity in different media, exhibiting not only an ultra-low OER overpotential of 266 mV at 10 mA/cm
2 but also a high-performance all-water decomposition over a wide pH range (
Figure 45)
[69][87].
Figure 45. XPS spectra of RuO
2–WC NPs, Ru–WC NPs and WC NPs. High-resolution XPS spectra of (
a) W 4f, (
b) Ru 3d and C 1s, and (
c) Ru 3p. (
d) Mechanism of OER process steps and corresponding Gibbs free energies of reaction intermediates on RuO
2–WC NPs. (
e) Free energy landscape of pristine RuO
2 and RuO
2–WC NPs at zero potential (
U = 0), equilibrium potential (
U = 1.23 V), and the potential (
U = 1.91 V) for which each step is downhill of RuO
2–WC NPs, respectively
[70][86].
3.4. Electron-Metal-Support Interactions (EMSI)
EMSI can lead to changes in the electronic structure of the metal particles and alter the adsorption and activation energies of reactant molecules, resulting in enhanced catalytic properties. In 2012, Campbell et al. first proposed electronic metal-support interactions (EMSI) based on the interaction between platinum and cerium atoms [71][88]. Specifically, EMSI refers to the charge redistribution phenomenon at the interface between the two, the support and the NPs, in a loaded metal nanoparticle catalyst. This is manifested as a local environmental change in the metal site, causing a change in the d-band (εd) structure of the metal. By designing and modulating EMSI, the εd of the metal can be increased, thereby enhancing the catalytic performance of the catalyst [67][71][84,88].
Catalyst systems with typical EMSI effects have been identified: metal-doped carbon (M-dN), metal-transition metal compound groups (M-TMCs) and metal-metal substrates [67][84]. EMSI involves the formation of chemical bonds due to the mixing of metal d-orbitals. On the one hand, this interaction stabilizes the metal NPs; on the other hand, the support effect will greatly influence the catalytic performance of the metal NPs [72][73][90,91].The energy balance at the metal-support interface reduces the activation energy of the reaction and thus increases the catalytic activity of the catalyst. The electron flow at the metal-support interface is decisive for the Fermi energy balance of the catalyst [74][92]. The properties of the support, the loaded metal NPs, defects, and so on can influence the electron transfer process [53][64]. The net electron transfer across the metal-support interface enhances the chemisorption of the metal NPs to the reactants [75][93].