Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Scott Kenney and Version 2 by Conner Chen.
In the 1980s, non-A, non-B hepatitis (NANB) was identified to be hepatitis E virus (HEV), and found to be the causative agent of large scale viral hepatitis outbreaks in developing countries. One of the distinctive epidemiological features of an HEV outbreak is its association with a high pregnancy mortality rate, potentially reaching up to 35% mortality. HEV is ow recognized as the leading cause of acute viral hepatitis worldwide. HEV is a 7.2 kb quasi-enveloped, single-stranded RNA virus with a positive-sense genome. HEV has been difficult to grow in cell culture necessitating animal models to understand pathology. Multiple animal models have been attempted to replicate pregnancy mortality and those trials are summarized herein.
- hepatitis E
- pregnancy models
- rabbit
1. Background
In the 1980s, non-A, non-B hepatitis (NANB) was identified to be hepatitis E virus (HEV) [1][28]. HEV was found to be 7.2 kb in size and a quasi-enveloped, single-stranded RNA virus with a positive-sense genome. The absence of peplomers at the membrane-associated HEV virions confirmed that the virus was quasi-enveloped, similar to the hepatitis A virus [2][29]. Three open reading frames (ORF 1, 2, and 3), edged by 5′ and 3′ untranslated regions (UTR), have been defined in the viral genome [3][30]. A 7-methylguanylate (m7G) cap is present at the end of the 5′ UTR, which drives cap-dependent genome translation. The 3′ UTR possesses a poly-adenylated tail [3][30]. Of the three conserved ORFs, ORF1 encodes for the non-structural polyprotein, which comprises functional domains that are essential for viral genome replication [4][5][31,32]. Bicistronic subgenomic RNA is responsible for the synthesis of overlapped ORF2 and ORF3 [6][33]. The viral capsid protein is encoded by ORF2 and possesses three domains: S (shell), M (middle), and P (protruding). Recently, ORF2 was shown to encode a secreted form (ORF2S), for which the biological role needed to be further elucidated [7][8][9][34,35,36]. ORF3 encodes a phosphoprotein that appears to play an important part during viral particle morphogenesis [10][37]. A fourth open reading frame has been reported in gt1 human and rodent HEV strains. Both ORF4s overlap ORF1 but contain little sequence conservation of the proteins between viruses. In human HEV, ORF4 appears to enhance replication during endoplasmic reticulum stress and, when ectopically expressed, enhances gt3 replication in cells, and the function of rodent HEV ORF4 is unknown [11][12][13][38,39,40].
2. HEV New Classification
HEV has recently undergone classification updates and continues to evolve with the discovery of new HEV strains. The recent reclassification of the family Hepeviridae comprises two subfamilies: Orthohepevirinae and Parahepevirinae. The former infects multiple terrestrial and arboreal animals that consist of four genera. Paslahepevirus balayani comprises eight genotypes (gt1–gt8), four of which currently cause the most cases of hepatitis E in humans (gt1, gt2, gt3, and gt4) [14][41]. HEV gt1 and HEV gt2 are obligate to humans, while HEV gt3 and HEV gt4 have zoonotic importance, and thus, the transmission occurs primarily from swine to humans via pig and undercooked pork products [15][42] but can also be transmitted by other species such as rabbits and deer. The three other genera, Avihepevirus, Rocahepevirus, and Chirohepevirus, largely circulate in birds, rodents, and bats, respectively.
3. Geographical Distribution of HEV
Although HEV is described as an emerging infectious agent, it is the established main cause of acute viral hepatitis (AVH) globally [16][43]. More than 20 million infections are anticipated to occur globally each year, resulting in nearly 70,000 infections leading to death [17][44]. Developing countries such as Asia, Africa, and Latin America are the primary regions that report major epidemic outbreaks resulting in deaths related to exposure to faecally-contaminated water that leads to outbreaks and sporadic cases of hepatitis E [17][18][44,45]. In addition, hepatitis E in these geographical regions is attributable to the human-associated HEV gt1 (Asia, Africa, and Latin America) and gt2 (sub-Saharan Africa and Mexico). However, the worldwide spread of zoonotic strains (gt3 and gt4) has lately been recognized as a reason for sporadic hepatitis in medically susceptible patients and the general population, including in high-income countries [19][20][21][22][23][46,47,48,49,50]. Several findings imply that hepatitis E is a zoonotic disease that is associated with pigs, rabbits, rats, and camels, which act as reservoirs for human infection [24][25][26][51,52,53].
4. Pregnancy Mortality Reported Due to HEV
Higher case-fatality ratios due to acute liver failure (ALF) in pregnant women in their third trimester remain a typical pathognomonic feature of hepatitis E outbreaks [27][28][29][30][54,55,56,57]. Alongside the fecal-oral route of transmission, parenteral and vertical routes of transmission have been described for HEV [31][32][33][58,59,60]. Limited data are available on the mother-to-child spread of HEV infection from India. It is reported that HEV infection produces serious liver disease with higher mortality rates in fetuses and neonates [34][61]. The survivors of vertically-transmitted HEV infection demonstrate self-limiting clinical progression with short-lasting viremia [34][61].
Evidence from 18th-century Europe demonstrates epidemic jaundice with an excess of illness and death among pregnant women and their infants, potentially representing the first historically documented accounts of HEV [28][35][55,62]. It was in the mid-1950s when the first retrospective study serologically validated a hepatitis E outbreak in Delhi, India; however, molecular proof hints that HEV may have already been flowing through humans for hundreds of years [3][29][36][37][38][30,56,63,64,65]. During the Delhi epidemic, the hospital-based study documented a 10% maternal case-fatality rate alongside miscarriage, stillbirth, or neonatal death in 56% of the infants of women with HEV infection. Notably, expiring and surviving infants were reported as having jaundice [39][66].
It has been over six decades since HEV was recognized as a reason for infectious hepatitis, and several subsequent studies have similarly reported high rates of maternal, fetal, and neonatal illness and death in pregnancies affected by HEV; however, the mechanism for pregnancy mortality has not yet been fully identified [40][67]. A report from epidemiological disease models has suggested that HEV might be responsible for almost 2400–3000 stillbirths per year in developing countries, with additional fetal deaths associated with antenatal maternal mortality [17][41][42][44,68,69]. Hepatitis E-related preterm delivery in mothers is common, with worse neonatal survival [41][43][68,70]. The reports from a 2002 outbreak in the Central African Republic indicated premature deliveries by all of the serologically confirmed hepatitis E (n = 7) pregnant women. Briefly, three stillborn cases, with one of them macerated and a fourth baby, were reported dead within a few minutes of delivery [44][71]. Similarly, in a 1993–1994 outbreak in Islamabad, Pakistan, all the newborns from acute hepatitis E mothers comprised 50% (4/8) of the fatalities [27][54]. During a 2008–2009 hepatitis E outbreak in Tongi, Bangladesh, pregnant women diagnosed with jaundice were more than two times as likely as non-jaundiced pregnant women to miscarry or deliver a stillborn baby [45][72]. In New Delhi and Chennai, India, two separate hospital-based prospective studies reported that 15–50% of the live-born infants of mothers with hepatitis E died within the first 1-week post-partum [41][46][68,73]. Likewise, the 2010–2011 outbreak in Sudan reported 14 intrauterine deaths and nine premature deliveries among 39 pregnant hepatitis E cases [47][74].
Similarly, in one report of eight infants born to HEV-infected mothers in their third trimester, seven infants were born at term, and one was born prematurely at 34 weeks gestation. Anti-HEV IgG was detected in all the infants, and HEV RNA was detected in five. One baby was jaundiced at birth with elevated serum ALT levels, and four were anicteric with elevated ALT levels. After birth, two babies developed hypothermia and hypoglycemia and died within 24 h. On autopsy and liver biopsy, one of these infants had massive necrosis [32][59]. Another report on nineteen infants born to HEV-infected mothers resulted in vertically transmitted HEV infection being seen in fifteen at birth, IgM anti-HEV positivity being detected in twelve, and HEV RNA being detected in ten, with three experiencing short-lasting IgG anti-HEV positivity because of maternal antibody spread across the placenta. Additionally, seven HEV-infected patients were reported with icteric hepatitis, five with anicteric hepatitis, and three with high serum bilirubin with normal liver enzymes. Seven infants died in the first week after birth, including one death due to prematurity. Of the nine survivors with follow-up data, five were HEV RNA positive. Interestingly, HEV RNA was not detectable in three patients by 4 weeks of life, in one by 8, and in the remaining infants by 32 weeks. The self-limited disease was seen in all survivors. They reported that HEV transmission from the mother to the fetus caused high neonatal mortality [34][61]. Therefore, the situation necessitates effective animal models for the study of HEV infection.
To date, there have been no animal models that could recapitulate all the clinical manifestations seen in humans (Figure 1). Rabbits, pigs, non-human primates, chickens, mice, rats, ferrets, and Mongolian gerbils have been used for pathogenesis, vertical transmission, and vaccine efficacy studies. However, none of them have been able to fulfill the characteristics that require them to be the ideal model for HEV pregnancy mortality.
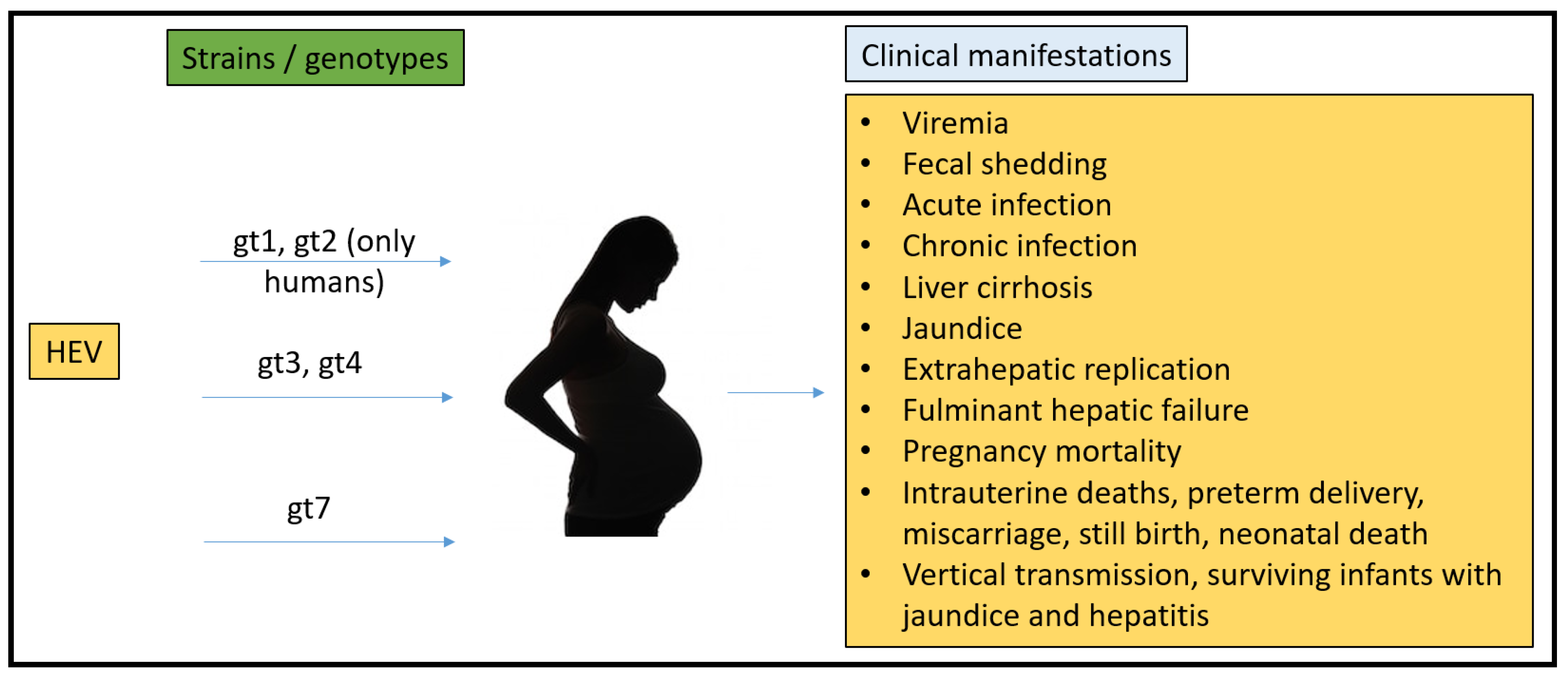
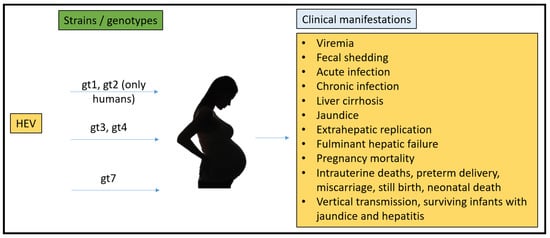
Figure 1.
Summary of HEV strains/genotypes and overall clinical manifestations in humans.