Acute disseminated encephalomyelitis (ADEM) is an immune-mediated central nervous system (CNS) disorder, characterized by polyfocal symptoms, encephalopathy and typical magnetic resonance imaging (MRI) findings, that especially affects young children. Advances in understanding CNS neuroimmune disorders as well as the association of myelin oligodendrocyte glycoprotein antibody (MOG-Ab) with both monophasic and recurrent forms of ADEM have led to new insights into its definition, management and outcome.
- acute disseminated encephalomyelitis (ADEM)
- acquired demyelination syndrome (ADS)
- myelin oligodendrocyte glycoprotein antibody (MOG-
- multiple sclerosis (MS)
- optic neuritis (ON)
- neuromyelitis optica spectrum disorder (NMOSD)
1. Brief Introduction and Historical Perspective
Acute disseminated encephalomyelitis (ADEM) is an inflammatory demyelinating disorder of the central nervous system (CNS) that predominantly affects children [1]. Whilst the first ADEM descriptions date back to the 18th century, the understanding of its physiopathology and definition have continued to evolve [2]. In the last decade, in an effort to characterize the range of acute demyelinating syndromes, the International Pediatric Multiple Sclerosis Study Group (IPMSSG) produced consensus clinical and radiologic diagnostic criteria defining ADEM as an acute demyelination syndrome that presents clinically with encephalopathy and polyfocal CNS symptoms showing demyelination on brain magnetic resonance imaging (MRI) [3][4][3,4]. As the imaging criteria were predicated on not having features characteristic of multiple sclerosis, challenges have arisen when utilizing imaging to diagnose ADEM [5] and predict other relapsing syndromes [6][7][6,7]. A key development in recent years is the recognition of the association of myelin oligodendrocyte glycoprotein antibody (MOG-Ab) with ADEM and recurrent forms of demyelination such as multiphasic ADEM (MDEM) and ADEM followed by recurrent optic neuritis (ADEM-ON) [8][9][1,8,9]. Insights into MOG-Ab-associated disease manifestation are starting to challenge older assumptions of ADEM [10][11][12][10–12]. Despite these advances, little change in acute therapeutic strategies has occurred, although the newer paradigm of timely initiation and optimization of immune treatments employed for a range of neuroinflammatory conditions is also beginning to be utilized for demyelinating disorders [13]. Protocols focus on early high-dose intravenous corticosteroids as first-line therapy and immunoglobulin and plasma exchange as second-line therapy with overall good outcomes based on observational studies [10][1,10]. The timing of treatment escalation and re-assessment of radiological findings remain unsolved questions. As for outcomes, which have historically been considered favorable, consistent findings have yet to shed light on the cognitive impairment reported in a few cohorts [14][15][1,14,15]. Here, we target this review on the clinical, epidemiological and pathophysiological aspects of pediatric ADEM, focusing on newer clinical perspectives.
2. ADEM in the context of ADS
Pediatric ADEM belongs to a group of disorders (Figure 1) characterized by acute or subacute onset of neurological deficits associated with evidence of inflammatory demyelination of the CNS, including the optic nerves, collectively named acquired demyelinating syndromes (ADSs) [16][17][4,16,17]. ADEM is a relatively common subtype of ADS, occurring in 22–32% of children with ADS [18][19][20][21][12,18–21].
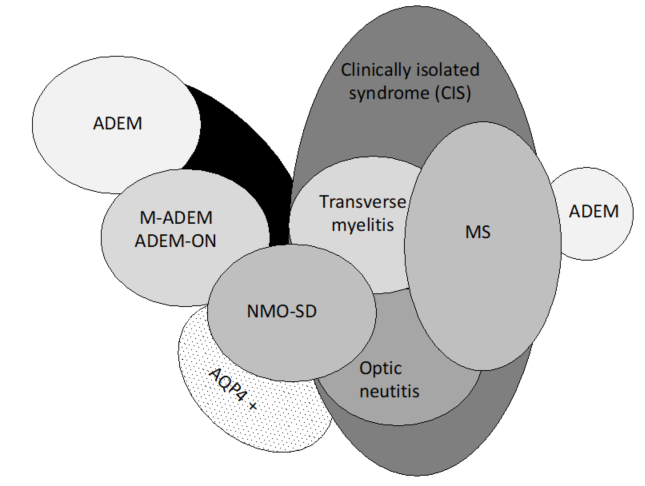
Figure 1. Spectrum of acquired demyelinating syndromes. ADEM, acute disseminated encephalomyelitis; MOG, myelin oligodendrocyte glycoprotein; AQP4 +, aquaporin-4; M-ADEM, multiphasic‐ADEM; ADEM-ON, ADEM followed by at least one optic neuritis; NMOSD, neuromyelitis optica spectrum disorder; MS, multiple sclerosis (adapted from Neuteboom et al. [16].).
Direct comparisons among earlier cohorts have been difficult because of varying definitions, as highlighted by Mikaeloff and Tardieu [22]. As such, differences across a range of many other demyelinating conditions catalyzed an international effort by the IPMSSG to develop diagnostic criteria [3,4]. The distinct subtypes of ADS are typically classified based on (1) clinical localization of symptoms and signs and (2) whether they are monophasic or relapsing diseases.
The first event of ADS in children remains monophasic in most cases and is mainly classified as ADEM or clinically isolated syndrome (CIS). By definition, ADEM requires the presence of encephalopathy and polyfocal CNS symptoms. As for CIS, the definition is applied for ADS presentations that can be either monofocal or polyfocal, reflecting clinical signs and symptoms in one or multiple CNS locations. Typical monofocal presentations are optic neuritis (ON), brainstem symptoms (i.e., internuclear ophthalmoplegia) and transverse myelitis (TM).
The first demyelinating event might also represent the first manifestation of a chronic demyelinating relapsing disease, such as multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD) or myelin oligodendrocyte glycoprotein antibody (MOG-Ab)-associated disease, recently recognized as a multiphasic disease course that does not resemble MS [23][24][23,24]. ADEM as a first manifestation of MS or NMOSD is uncommon <10% [25][1,25]. Typical brain MRI characteristics and aquaporin-4 antibody (AQP4-Ab), respectively, help to distinguish these disorders [9]. This process of diagnostic refinement is still ongoing, illustrating the need for further biomarkers.
The IPMSSG definitions for monophasic ADEM and relapsing disorders are shown in Table 1.
Table 1. Criteria for acute disseminated encephalomyelitis (ADEM) and relapsing disorders following ADEM.
|
ADEM |
Single polyfocal clinical CNS event with presumed inflammatory cause Encephalopathy that cannot be explained by fever MRI typically shows diffuse, poorly demarcated, large >1–2 cm lesions predominantly involving cerebral white matter; T1 hypointense white matter lesions are rare; deep gray matter lesions (e.g., thalamus or basal ganglia) can be present No new symptoms, signs or MRI findings after three months of initial ADEM |
|
Multiphasic ADEM |
New event of ADEM three months or more after initial event that can be associated with new or re-emergence of prior clinical and MRI findings |
|
ADEM-ON |
At least one subsequent attack of optic neuritis, without encephalopathy, at least three months after initial ADEM |
|
ADEM-MS |
ADEM followed three months later by a non-encephalopathic clinical event with new lesions on brain MRI consistent with MS |
|
ADEM-NMOSD |
ADEM followed three months later by ON, myelitis or area postrema syndrome, fulfilling NMOSD diagnostic criteria |
ADEM, acute disseminated encephalomyelitis; CNS, central nervous system; ON, optic neuritis; MRI, magnetic resonance imaging; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorder. Adapted from Pohl et al. [1].
3. Epidemiology
Although it can afflict people at any age, ADEM is more common in children, with a median age at presentation of 5–8 years [26]. A male preponderance had been shown in most studies, with a male:female ratio ranging from 1:0.8 to 2.3:1 [27].
Reported annual incidence varies from 0.07–0.9/100,000 children in different locations [27][28][29][30][1,7,17,27–30]. It has been described throughout the world and affects all ethnicities, but its incidence is higher with increasing distance from the Equator, similar to the geographic distribution of MS [31].
Mechanistically, ADEM has been classified as a predominately post-infectious CNS disorder, with an identifiable trigger reported in up to 50–85% of cases [32][28,32]. First ADEM descriptions were commonly associated with infections such as smallpox and measles, with high mortality and neurological sequelae. Although immunization programs reduced the incidence of ADEM related to those infections, ADEM continues to be associated with other infections, especially, but not exclusively, viral infections. Neurological symptoms generally begin within 2–21 days (range 1–42) after an infection, and the most frequent are flu-like symptoms, upper respiratory tract infection and gastroenteritis. Bacterial infections have also been implicated [26,28]. Although some studies [32] required previous infection for ADEM diagnosis, this was not included in the current diagnostic criteria.
Although vaccinations have also been implicated as a cause of ADEM, no clear pathogenetic correlation exists and the incidence of ADS following infection is higher than that induced by immunization itself [33][1,33,34]. The frequency of ADEM occurring after vaccination has decreased in recent years, probably due to changes in the methods used to produce vaccines [33]. Almost all immunizations have been implicated, especially rabies, hepatitis B, polio, influenza, pertussis, measles, mumps and rubella [26]. A first vaccination is more associated with ADEM than re-vaccination [33]. A recent large case–control study found no association of vaccines with ADS in a three-year follow-up period of both children and adults [34]. This finding emphasizes that vaccines might have a similar effect as infections in triggering the first episode of ADS and may accelerate the transition from subclinical to relapsing ADS in patients with an existing risk for autoimmune disease.
4. Clinical and Radiological Perspective
4.1. Diagnostic Criteria
The diagnosis of ADEM is based on a combination of clinical features, supported by MRI findings (see Table 1) [4]. Importantly, other diseases need to be excluded before a definite diagnosis can be made. Based on the most recent IPMSSG criteria, ADEM is characterized by polyfocal neurological deficits. Notably, the presence of encephalopathy is an obligatory feature. The definition of encephalopathy encompasses behavioral changes and/or alterations in consciousness, including irritability, not explained by systemic febrile illness or post-ictal symptoms. The obligatory presence of encephalopathy has been debated but remains present in the criteria, mainly due to the lack of sensitive and specific MRI criteria, along with serum or CSF (cerebrospinal fluid) biomarkers [35][36][21,35,36]. As there may be fluctuations in the disease course in the first three months, i.e., treatment-related fluctuations caused by cessation or tapering of steroids, any worsening, recurring or new symptoms within three months are still attributed to the first event. If there is a second event after three months that again qualifies as ADEM, the term multiphasic ADEM is used. The term recurrent ADEM, suggested in the 2007 IPMSSG criteria, was omitted in the 2012 IPMSSG criteria due to its low frequency in reported studies [3,4]. Another multiphasic phenotype is ADEM followed by (recurrent) optic neuritis (ADEM-ON). A diagnosis of MS after a first event of ADEM is rare, but possible if a child with ADEM experiences two subsequent non-encephalopathic events or one new event with the appearance of new MS-specific MRI lesions that are dispersed across time and space.
4.2. Clinical Features
A preceding infection or illness is observed in 70–80% of cases [37]. Although often preceded by a prodromal phase (malaise, headache), disease onset is subacute to acute. Progression of the disease usually takes places within days. Neurological signs include pyramidal signs, ataxia, brainstem symptoms, optica neuritis and transverse myelitis [37][38][1,37,38]. Symptoms may also include atypical signs like meningism, fever and seizures, resembling infectious meningo-encephalitis. Intensive care unit (ICU) management is reported for 15% of children with ADEM [38]. Clinical recovery typically occurs over weeks after the peak. Rarely, systemic involvement can occur, including cardiac complications with myocardial tissue damage [39][40][39,40].
4.3. MRI Findings
Brain MRI in the acute stage shows hyperintense abnormalities in T2-weighted and fluid-attenuated inversion recovery (FLAIR) images [4]. Lesions typically are bilateral, asymmetrical, large (>2 cm) and poorly demarcated. Both white and gray matter can be affected. Cortical as well as deep gray matter lesions have been described. Figure 2 shows some examples of typical ADEM MRI features. Gadolinium enhancement is not a typical feature of ADEM, reported in up to 30% of cases [14]. The frequency of spinal cord abnormalities is not well known, as spinal cord imaging is not routinely done in all ADEM cohort studies. If present, spinal cord abnormalities can be observed over more than two vertebral segments [14,35,38]. Complete resolution of MRI abnormalities has not been studied systematically, likely due to the need for sedation of young children for the procedure. This may be the reason for conflicting data on the resolution of MRI abnormalities [41][42][14,41,42].
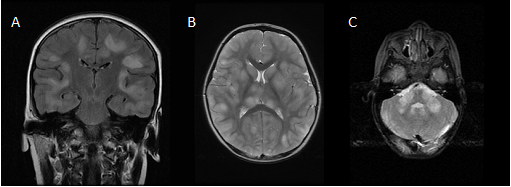
Figure 2. Typical magnetic resonance imaging features of ADEM: (a) coronal fluid-attenuated inversion recovery (FLAIR) image showing asymmetrical bilateral subcortical white matter abnormalities; (b) axial T2-weighted image depicting asymmetrical bilateral white and gray matter abnormalities; (c) axial T2-weighted image showing typical cerebellar peduncle lesions.
Notably, MRI may show no abnormalities in the first days [42]. Additionally, during clinical recovery, MRI imaging may still show worsening, indicating a lag between clinical symptoms and MRI abnormalities. As some patients with ADEM may have a multiphasic disease course and even develop MS, a reference scan timed around three months after ADEM onset is advised.
In order to discriminate between ADEM and MS at the time of the first attack, three items are potentially useful: (1) two or more periventricular lesions, (2) presence of black holes and (3) absence of a bilateral lesion pattern. If two of these three items are present, MS is more likely than ADEM. Yet, it should be noted that these criteria are not diagnostic, and the last item especially can be variably interpreted [43][44][43,44].
4.4. Laboratory Findings
Blood investigations show leucocytosis or elevated C-reactive protein (CRP) or erythrocyte sedimentation rate (ESR) in around 50% of children with ADEM, and therefore do not discriminate between ADEM and infectious meningo-encephalitis [38]. Data on cerebrospinal fluid (CSF) findings differ between studies. CSF pleocytosis is observed in a wide range of patients (28–86%) [45][14,35,38,45]. There is no strict upper limit to the pleocytosis, although CSF leukocyte cell counts above 100/μL are rarely seen and warrant against the possibility of an infectious meningoencephalitis [38]. Elevated CSF protein levels are observed in 23–66% of cases [45][46][14,35,45,46]. This may be an underestimation, as the normal values of CSF proteins in children are lower than in adults [46]. Oligoclonal bands are found in less than 10% and may be transitory, contrary to MS [45][14,35,38,45].
4.5. Differential Diagnosis
The differential diagnosis of ADEM is broad and contains, besides other ADSs, other inflammatory diseases and vascular, metabolic and genetic disorders [47][1,47]. Stroke-like events should point towards CNS vasculitis, systemic lupus erythematosus (SLE), Behcet’s disease, antiphospholipid antibody syndrome or mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELASs). Persistent seizures or predominant extrapyramidal movement disorders like dystonia and chorea may be symptoms of anti-N-methyl-D-asparate receptor (anti-NMDAR) encephalitis. This situation generally shows normal MRI, although white matter involvement has been described [47][13,47]. Bilateral symmetrical lesion patterns should guide the clinician towards diseases such as primary or secondary hemophagocytic lympho-histiocytosis and/or genetic/metabolic disorders and leukodystrophies. A progressive disease course is another red flag and is indicative of genetic/metabolic disorders or neoplastic diseases like gliomatosis cerebri or lymphoma.
4.6. Comparison between Children and Adults
Making comparisons between children and adults with ADEM is difficult, as there are no international definitions of ADEM in adults. Although the clinical presentation in children and adults is comparable, there is evidence that the disease course and outcomes are worse in adults than in children [38]. Adults have a higher frequency of ICU admission, longer hospitalization, poorer recovery and higher mortality [38]. In children, MRI abnormalities more often show thalamic and basal ganglia lesions, whereas adults more often show periventricular lesions [35,38] and higher Gad–enhancing lesions (30% vs. 55–60%). A multiphasic, non-MS disease course has been reported in both children and adults.
4.7 MOG in Monophasic and Relapsing Forms of ADEM
MOG-Abs were recently detected in a subgroup of children with ADS, in particular in children with a non-MS like disease course [48][49][50][23, 24, 48]. The expression of serum MOG-Ab is age dependent and associated with different disease manifestations, including ADEM, ON and TM, either alone or in combination [48]. In young children, ADEM is the predominant clinical manifestation, whereas older children with MOG-Ab present with ON, myelitis or brainstem symptoms. The clinical course of children with ADEM and MOG-Ab is primarily characterized by encephalopathy in addition to polyfocal neurological signs. Imaging of the brain and spine shows widespread involvement of different anatomical areas including the brainstem and spinal cord, often with longitudinally extensive transverse myelitis (LETM) [51]. Children whose MOG-Ab level declines to undetectable levels are less likely to have further relapses and more likely to have a favorable long-term prognosis [51]. Children with a sole monophasic ADEM or subsequent recurring demyelinating events are difficult to distinguish on clinical and radiological grounds [52]. Standard laboratory findings for children with a relapsing course, such as CSF cell count and the presence of oligoclonal bands (OCBs), are comparable to those for children with a monophasic disease course [53]. There are three subgroups of children The clinical course of children with ADEM and MOG-Ab is primarily characterized by encephalopathy in addition to polyfocal neurological signs. Imaging of the brain and spine shows widespread involvement of different anatomical areas including the brainstem and spinal cord, often with longitudinally extensive transverse myelitis (LETM) [51]. Children whose MOG-Ab level declines to undetectable levels are less likely to have further relapses and more likely to have a favorable long-term prognosis [51]. Children with a sole monophasic ADEM or subsequent recurring demyelinating events are difficult to distinguish on clinical and radiological grounds [52]. Standard laboratory findings for children with a relapsing course, such as CSF cell count and the presence of oligoclonal bands (OCBs), are comparable to those for children with a monophasic disease course [24, 48, 53]. There are three subgroups of children with further demyelinating episodes following ADEM.
4.7.1. MDEM
According to the last IPMSSG report, MDEM is defined as an ADEM attack followed >3 months later by a second ADEM episode without any further attacks [1, 4]. Nevertheless, some patients present with more than two ADEM attacks in combination with persistent MOG-Ab [51][10, 12, 51]. The second demyelinating event generally occurs in the following 12 months, but the time interval and frequency of attacks vary considerably among patients, and can take up to four years for the second event [48]. These children develop new clinical symptoms with encephalopathy and focal neurological signs and new MRI findings characterized by hazy, large and bilateral widespread lesions [51][48, 51].
4.7.2. ADEM-ON
These patients have frequent attacks of mainly unilateral inflammation of the optic nerves, ranging from one to nine episodes, occasionally in combination with further ADEM-like attacks[53] [8, 53]. Interestingly, children during the recurrent ON phase rarely show new MRI lesions.
4.7.3. ADEM-NMOSD
A third group of children with MOG-Ab have further demyelinating episodes characterized by LETM and ON either simultaneously or, more often, sequentially, thus fulfilling the diagnostic criterion of AQP4-Ab-negative NMOSD. These children show a wide range of MRI findings which can be indistinguishable from children with AQP4-Ab. As in children with MDEM or ADEM-ON, serum MOG-Ab titers remain high over time.
5. Treatment
5.1. Acute Phase Treatment
As there are no specific randomized trials for ADEM, treatment protocols are derived from observational studies and expert opinions. Supportive care is important, and treatment with antivirals and antibiotics is generally prescribed, as ADEM may mimic infection [26, 67]. Once a diagnosis of ADEM is suspected, early treatment may contribute to a better outcome compared to historic reports [1, 13, 28]. First-line acute treatment generally consists of IV methylprednisolone at a dose of 30 mg/kg/day (maximum 1000 mg/day) for 3–5 days, followed by an oral prednisone taper for 4–6 weeks [1, 67]. Early discontinuation of steroids (<3 weeks) can increase the risk of relapse [68]. Steroid treatment requires close monitoring of blood pressure, electrolytes and glucose and administration of gastric protection [69]. Intravenous immunoglobulin (IVIG) is prescribed as second-line treatment for steroid-unresponsive ADEM at a total dose of 2 g/kg for 2–5 days. IVIG is generally well tolerated in children [70]. The rational use of IVIG is shown in patients with recurrent or steroid-dependent demyelination [26]. An ongoing trial of early IVIG in children with encephalitis might help in reconsidering the position of IVIG in brain inflammation [71]. Plasma exchange (PLEX) with three to seven exchanges is used in refractory patients [26, 32, 67, 72-75]. The usefulness of PLEX has been shown in trials of adults with demyelinating disorders [73] and in patients with AQP4-ab NMOSD as early therapy [74, 76]. Recent pediatric studies advocate PLEX as a safe and effective rescue therapy for inflammatory CNS disorders, including ADEM [72, 75]. Craniectomy has been performed in fulminant cases with increased intracranial pressure unresponsive to immunotherapy and critical care measures [69].
Recovery is expected within days after the initiation of therapy [1, 26]. A plan for follow-up MRI is required to assess multiphasic disorders [67]. Although radiological frequency remains controversial, it might be reasonable to wait 3 months from the event, as radiologic findings can fluctuate in this time period.
5.2. Relapsing Forms
Most patients will have monophasic disease, whereas 10–36% will have another demyelinating event [52, 67]. Patients diagnosed with MS or NMOSD after an ADEM episode are treated with standard disease protocols.
Therapy for recurrent MOG-Ab-associated diseases remains a challenge. In a recent European collaborative study, it was shown that disease-modifying treatments such as interferons or natalizumab, commonly used for MS patients, were not associated with clinical improvement in children with MOG-Ab-associated disease, whilst B cell-targeted treatments, particularly intravenous immunoglobulins, were associated with a reduction in relapse frequency [11]. Other reports suggested that continuous oral steroid treatment might be associated with a reduced relapse rate. Further studies are needed to address the optimal treatment and outcome in this group of children.
6. Outcome
Approximately a quarter of all children hospitalized with ADEM require admission to the intensive care unit [77]. The mortality rate was reported as 1–3% [1, 77]. Even though there is no uniform evaluation among different ADEM studies, full recovery with normal neurological examination is reported for most patients (50–80%) [67, 70]. Long-term motor deficits, visual problems and seizures are rare [14]. Distinct cognitive problems are reported in up to 56% of patients [15, 67, 69]. Recent pediatric MOG-Ab studies found that poor cognitive outcome was associated with the ADEM or ADEM-ON phenotype [11, 51, 78] at a higher frequency compared to adult studies [8]. Academic disorders as an indirect measure of poor cognition were also associated with ADEM and younger age at onset and deep gray matter lesions on MRI scans [79]. A recent evaluation of children with acquired demyelination demonstrated reduced brain volume and reduced expected brain growth even in the absence of chronicity, especially in 18 ADEM patients. It is postulated that a higher volume of white matter lesions in ADEM patients and their younger age at onset might be responsible for the lack of expected white matter growth in these patients [54][80].