2. CDs’ Optical
Optical Properties
The most interesting and appealing characteristic of CDs is the great intensity of their fluorescence emission
[34][62] that originates from quantum confinement (QC) occurring when the exciton’s Bohr radius is bigger than the average size of CDs
[35][63]. The nanometer size of carbon dots does not allow the formation of crystal-like conduction, and the valence bands and the electronic level are discrete although somewhat broadened. The HOMO–LUMO gap increases when the CD size decreases, leading to the emission of photons in the UV region with an improvement in the quantum yield (QY)
[36][64]. The fluorescence emission for CDs with π-domains of larger size (i.e., GQDs, CQNDs) is mainly due to conjugated π-electrons of the aromatic carbon domains
[37][65]. Larger π-domains reduced the HOMO–LUMO gap and red shifted the fluorescence emission peak
[34][62]. The same consideration holds for all xGQDs, although some adjustments are needed because of the presence of different heteroatoms.
Considering CQNDs, Yu et al.
[38][66] investigated another interesting effect on fluorescence coming from the core/shell size ratio and surface residues suggesting the fluorescence emission mechanism reported in
Figure 1.
Figure 1. Scheme of the fluorescence emission of CQNDs based on different core/shell size ratios and surface residues.
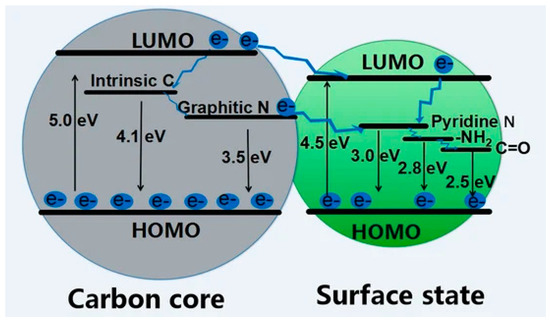
CDs’ core-related fluorescence is not the only emission mechanism as surface states also play a role. The great complexity of surface defects and related states is reflected by the various simultaneously active mechanisms such as excitation-dependent luminescence and pleochroism. As reported by Yan et al.
[34][62], shell surface defects are capture centers for excitons and promote radiative relaxation from excited states to the ground state, leading to multicolor emissions, the red-shift being determined by the oxidation degree
[39][67]. Du et al.
[40][68] suggested that shell functional groups bonded to the edge of honeycomb carbon fragments (i.e., hydroxyl, carbonyl, carboxylic residues and their heteroatom derivative) are at the origin of the visible fluorescence in xGCDs. The authors hypothesized that amides play a major role in the blue emission while carboxylic derivatives induce a redshifted emission.
CPDs fluorescence is closely related to the shell surface fluorescence of xGQDs due to the absence of a proper graphitic core. Accordingly, the aromatic clusters are dispersed and connected to each other through sp
3 orbitals and are deemed the source for fluorescence emission
[41][69]. As a matter of fact, the energy levels and electron cloud distribution of π and π* states in the aromatic domains could be influenced by their interaction with σ and σ* states of sp
3 carbon surrounding
[42][46]. Nevertheless, the great variability of CPDs requires a more detailed discussion that also considers structural and chemical features to provide a comprehensive picture of fluorescence emission. CPD fluorescence could arise from fluorophore residues retained or formed during the synthesis
[43][70] of a heteroatoms rich regions. As reported by Qu et al.
[44][71], nitrogen-enriched CPDs showed an increment of QY while other elements such as sulphur
[45][72], boron
[46][73] or fluoride
[47][74] could increase the HOMO–LUMO gap, enhance the charge transfer or stabilize the fluorescence emission in a wide pH range, respectively. Nonetheless, CPDs could be fluorescent even without the presence of aromatic domains due to the crosslink enhanced emission effect. This phenomenon was firstly described by Tao et al.
[48][75] by comparing the fluorescence emission of poly(ethylenimine) with CPDs-derived materials produced by the degradation and crosslinking of the polymer. The authors reported a magnification of fluorescence emission and they suggested that this was due to the formation of a rigid structure able to forbid the intramolecular rotations promoting the fluorescence emission. This was supported by further studies, indicating that the mobility reduction of the covalent bond due to steric hindrance or supramolecular interactions could further improve the fluorescence of CPDs
[49][50][76,77].
Several mechanisms of CD chemiluminescence constitute the pillars of numerous different applications. CD fluorescence could be used for two main different applications, one based on its quenching and one based on its magnification.
CD fluorescence quenching represents a profitable analytical tool for the detection of metals
[51][52][78,79] and organic species
[53][80]. The mechanisms of fluorescence quenching are multiple and are related to the interactions between CDs and the quencher molecules. The quenching mechanisms are (i) dynamic quenching
[54][81], (ii) static quenching
[55][82], (iii) photoinduced electron transfer
[56][83], (iv) Förster resonance energy transfer
[57][84], (v) Dexter energy transfer
[58][85], (vi) inner filter effect
[59][86] and surface energy transfer
[60][87].
The dynamic fluorescence quenching is due to a diffusion quencher while CDs are in their excited state. The fluorescence dynamic quenching of CDs is temperature-sensitive and can be used for the detection of both inorganics and organic compounds
[61][88]. Gonçalves et al.
[62][89] attributed the CDs’ quenching observed in the presence of Hg(II) to the dynamic quenching effect due to the intensity of the phenomena without supporting it with more detailed investigations. A similar consideration was reported by Wang et al.
[63][90], suggesting that the simultaneous dynamic and static fluorescence quenching. Contrary to dynamic one, static fluorescence quenching occurs through the formation of a ground state complex between the quencher and CDs
[64][91]. The static quenching of CD fluorescence is tentatively exploited for the detection of inorganics that are forming stable complex species with CDs. Xu et al.
[65][92] reported static quenching for poly(ethylene glycol)(PEG)-coated CPDs in the presence of Pt(IV) and Au(III). The metal–shell interactions promoted a great quenching of native fluorescence emission intensity without altering the fluorescence decay pathways. Authors suggested that it was due to static quenching involving the absence of electron–hole radiative recombination. Alternatively, to monitoring fluoresce quenching, Gong et al.
[66][93] prepared quenched CPDs by creating stable complexes with Fe(III). The quenched CPDs were used to detect ascorbic acid due the formation of iron ascorbate, removing the quencher from CPDs with a corresponding enhancement of fluorescence emission. Wang et al.
[67][94] proved the ability of CQNDs to form stable complexes on the shell with several metal ions through complex formation with nitrogen-based residues. The interactions between metals and CDs could also be destructive as reported by Song et al.
[68][95]. The authors showed that, in presence of oxygenated water, Fe(II) could promote a Fenton reaction with the chemical oxidation of CDs.
Photoinduced electron transfer quenching occurs on distances greater than 10 nm between the quencher and CDs
[69][96], while the Förster resonance energy transfer quenching takes place for distances lower than 10 nm and involves dipole–dipole interactions
[70][97]. These three phenomena were investigated as possible new routes to convert photons into electrons for the production of molecular devices
[71][72][98,99]. Nonetheless, Förster resonance energy transfer has found an interesting application in the development of a dual species probe composed by CDs and metals clusters
[73][100]. This approach allowed to quench CD fluorescence and then reactivate it when metal clusters are removed by the presence of analytes.
The distance between the CDs and the fluorescence-quenching mechanism is also affected by the condition of the CD system. Yoo and co-workers
[74][101] investigated the effect of dilution on GQDs with a controlled carbonization degree. As stated above, the fluorescence emission of GQDs is mainly due to π-conjugated domain of the core but the surface states play a relevant role in promoting the quenching aggregated state. This dual role of the π-conjugated domain in CDs offers a challenge to the prediction of photoluminescence (PL) characteristics in the solid-state. By tuning the core–shell ratio and the aromatic domains size, the authors proved that the CDs with a higher core–shell ratio of π-conjugated domain are dominated by a core emission in the concentrated solution with the total suppression of surface emission. Dexter energy transfer quenching occurs on a similar scale but it is based on orbital overlap
[75][102]. The inner filter effect was originally considered as an error in fluorescence measurements, but it was not. It is a particular phenomenon due to the overlap between the excitation or emission spectrum of CDs and the absorption spectrum of quencher
[76][103]. Surface energy transfer quenching takes place more in quantum dots that in CDs and it is due to interactions between surface plasmons and the orbital system of a fluorophore
[77][104]. Nonetheless, it can also occur between CDs and small metal clusters
[78][105].
An interesting phenomenon related to CD fluorescence is the excitation-dependent emission and its origin is debated in several studies
[79][80][106,107]. A relevant study was conducted by Krishnaiah et al.
[81][108] using a top–down approach to hydrothermally convert blue grass into CPDs. Authors reported the remarkable redshift of a fluorescence emission peak from 370 to 470 nm using a wavelength ranging from 280 up to 400 nm. The authors hypothesized that the red shift occurred due to n→π* transitions between non-bonding molecular orbitals of carbonyl and carboxylic groups on the CPDs shell. This was in good agreement with the modelling of PCDs’ structures reported by Mintz et al.
[82][109] and with the nitrogen doping detected. The performances of PCDs were reasonable, with a quantum yield of 7% and the ability of detecting of Mn(II) and Fe(III). Surprisingly, the authors detected a loss of fluorescence quenching in the presence of both Pb(II) and Cd(II) suggesting the poor complexability of these cations by the functionality residues of the shell. The excitation-dependent emission is rather challenging due to the violation of the Kasha–Vavilov rule
[83][110] and a first attempt to provide a theoretical explanation was provided by Khan et al.
[84][111]. The authors investigated the fluorescence of CQNDs by a nanosecond time resolving spectroscopy reporting a significant energy redistribution and a relaxation among the emitting states. The authors proved that the inhomogeneous broadening and red shift of the emission peak were due to a system of energy sub-states and not caused by a quantum confinement of a different-sized CQNDs
[85][112] and only weakly related to the oxidation of shell edges. This last point is still debated and the other authors suggested that the red shift was mainly related to the shell oxidation degree
[86][113]. Experimental results are inconclusive and we believe that both interpretations are possible even if they reached opposite conclusions. This is related to the variability in CQNDs structures and unique features that make it impossible to provide a unique explanation for the emission-dependent red shift and require a two-limit scenario as mentioned above, which is able to describe all the CQND materials produced using different routes.
CDs could also show another photoluminescence mechanism: phosphorescence
[7]. Phosphorescence originates from the intersystem crossing from the lowest excited singlet state level to the triplet state and from radiative decay from the lowest excited triplet state to the ground state one. Particularly, PCDs are quite effective to exploit phosphorescence due to the crosslinking between aromatic domains and supramolecular interactions, RTP can be easily achieved through appropriate design without additional matrices, due to covalently cross-linked frameworks, polymer chains and supramolecular interactions
[87][114]. Tao et al.
[88][115] systematically studied the PCD phosphorescence, showing that the crosslinked matrix suppressed the nonradiative transitions due the covalent bonding of the neighboring emissive centers. Accordingly, the energy level distributions reduced the energy gap compared with the precursors promoting the formation of triplet states. Furthermore, the complex network of hydrogen bonds contributed to the system rigidity, decreasing the nonradiative relaxation. Phosphorescence was also observed in xGQDs: Song et al.
[89][116] exploited the UV phosphorescence of CQNDs by reducing the graphitic domains or reducing the mobility of CD domains by adding sodium isocyanate crystals. The authors proved that the electron transition from the p
x to the sp
2 orbital of the nitrogen generated an orbital angular momentum able to populate the triplet state. At the same time, the encapsulation of CQNDs reduced the energy dissipation triplet excitons reaching a phosphorescence lifetime of up to 16 ms. Similar results could be achieved by entrapping CQNDs into the silica
[90][117] or polymeric matrix
[91][118]. Knoblauch et al.
[92][119] followed a different route to achieve phosphorescence CDs based on tailoring with bromine. Bromine-doped CDs had accessible triplet states exploiting the phosphorescence in liquid media at room temperature. A key factor that must be considered for the preparation of phosphorescent CDs is the stability. As discussed in the review work of Wei et al.
[93][120], long exposure to UV light induced a loss of shell residues leading to the quenching of phosphorescence. Hu et al.
[94][121] faced this issue by tailoring the CD surface with silicon residues, prolonging the lifetime under hard UV light up to 200 h.
Another relevant optical property of CDs is their chemiluminescence
[95][122]. Chemiluminescence is produced during chemical reactions in which intermediate radical species decompose to form electronically excited species and deactivate themselves. CD chemiluminescence originates from the fluorophore residues that act as emitting centers, as first reported by Lin et al.
[96][123]. CD chemiluminescence has found plenty of applications in optoelectronics and catalytic processes related to the various structures of CDs available with unique fluorophores and energy levels
[97][124]. Interestingly, Xu et al.
[98][125] reported the tunability of chemiluminescence response by tuning the oxidation degree of CDs. The authors proved that highly oxidized CDs showed a high fluorescence emission with chemiluminescence while the opposite was true for poorly oxidized CDs. The authors concluded that fluorescence was related to the core states for photon absorption with the shell acting as a long wavelength trap providing nonradiative recombination centers. Conversely, chemiluminescence was related to the radicals formed in the shell surface states.