Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yulia Volkova and Version 2 by Catherine Yang.
Phosphorus(V)-substituted pyridine was first synthesized by Plazek’s research group in 1936 by a reaction of 2-dimethylaminopyridine with phosphorus trichloride under oxidative conditions. Later, related compounds were obtained by reactions of metalated pyridines with phosphorus-halogen compounds, of pyridinediazonium tetrafluoroborate with phosphorus trichloride, of N-alkoxypyridines with sodium diethyl phosphite and phosphines, and of N-pyridylpyridines with phosphonic acid, by the Michaelis–Arbuzov reaction, Pd(II)-catalyzed phosphorylation of halopyridines, cyclization of phosphorus-containing 3-azatrienes, and the Diels–Alder reaction involving 3-phosphoryl-1-azadienes.
- heterocycles
- pyridine
- Michael reaction
- phosphorylation
- carbenoid-mediated reactions
- formal cycloaddition
1. Cyclizations Based on the Michael Reaction
In recent years, synthesis of pyridines via the Michael reaction using phosphoryl-substituted Michael acceptors and donors has gained significant attention. Allais et al. [1][2][3][63,64,65] described three-component condensation of 1,3-dicarbonyl compounds 3 with P(O)Et2-bearing vinyl ketones 2 and ammonium acetate (1) in the presence of oxygen, leading to pyridine-2-phosphonates 4 (Scheme 1). The authors suggested that the reaction occurs through successive addition of compound 3 to Michael acceptors 2, providing ketone 5 followed by enamination of the latter with ammonia (1′) to form intermediate 6, which undergoes intramolecular cyclization into dihydropyridine 7. Complete oxidation of intermediate 7 to pyridine 4 was achieved using oxygen in the presence of activated carbon. The reaction was general with respect to β-oxo esters and β-oxoamides, providing products in 49–80% yields.
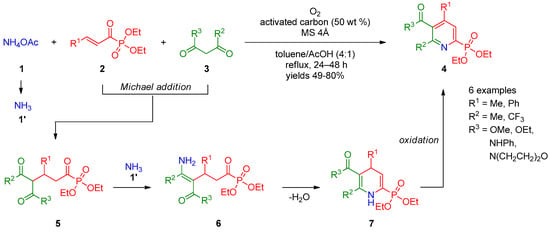
Scheme 1.
Synthesis of pyridine-2-phosphonates.
Hanashalshahaby and Unaleroglue [4][66] showed that pyridine-3-phosphonates 10 can be obtained by three-component oxidative coupling of diethyl (2-oxobutyl)phosphonate (8) with Mannich bases 9 and ammonium acetate (1) in the presence of catalyst K-10 (Scheme 2). The product yields are reasonable both for aryl- and alkyl-substituted Mannich bases. The authors hypothesized that β-keto phosphonate 8 reacts with ammonia 1′ generated in situ from ammonium acetate (1) to form enamine 11, which is accompanied by thermal decomposition of Mannich base 9, giving α,β-unsaturated carbonyl compound 9′. These two intermediates are subjected to the Michael addition to form ketoamine 12, which undergoes intramolecular cyclization to dihydropyridine 13, followed by oxidation of the latter with atmospheric oxygen to provide final product 10.
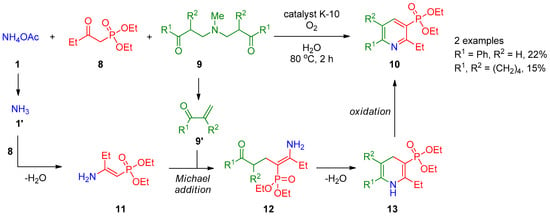
Scheme 2.
Reaction of diethyl (2-oxobutyl)phosphonate with Mannich bases.
Further, Abdou et al. [5][67] found that diethyl (2-amino-2-thioxoethyl)phosphonate acts as an efficient Michael donor in the addition reaction with β-(dimethylamino)vinyl ketone, thus leading to 2-thioxopyridin-3-ylphosphonate. Liao et al. [6][68] described one example of the aza-Michael reaction between diethyl (3-phenyl-3-oxopropyn-1-yl)phosphonate with methyl 3-aminocrotonate, providing phosphonate-ester-containing pyridine moiety under mild conditions.
2. Carbenoid-Mediated Reactions
Recently, several studies by Park and co-workers demonstrated the prospects of using metal carbenoids in synthesis of phosphoryl-substituted pyridines. They described [7][69] Rh(II)-catalyzed intramolecular cyclization of δ-diazo oximes to pyridines and showed that this is a facile method for synthesis of pyridine-2-phosphonate 15 (Scheme 3). Rh2(CF3CONH)4 was used as the catalyst of choice. The authors proposed a mechanism that involves reaction of the diazo group of oxime 14 with Rh(II), providing rhodium carbenoid 16, followed by insertion of the latter into the N–O bond to form dihydropyridinone 17, which undergoes aromatization to final pyridine 15 via elimination of methanol.
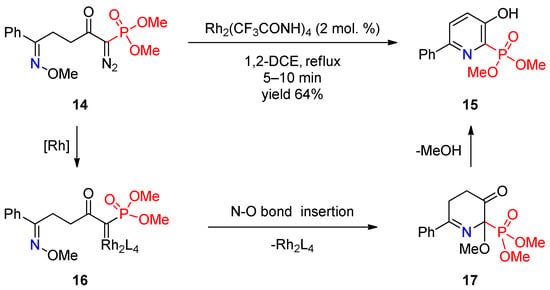
Scheme 3.
Rh(II)-catalyzed cyclization of δ-diazo oxime ether.
In another work by Park and co-workers [8][70], synthesis of pyridine-2-phosphonate 20 was accomplished using phosphorylated vinyl carbenoid 21 generated in situ from diazophosphonate 19 and dirhodium(II) catalyst Rh2(esp)2 (Scheme 4). Addition of compound 21 to 2H-azirine 18 affords intermediate 22. The latter undergoes three-membered ring opening accompanied by elimination of the Rh(II) catalyst to provide 3-azahexatriene 23, followed by cyclization to yield pyridine 20.
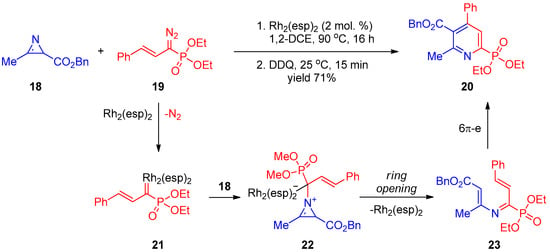
Scheme 4.
Reaction of phosphorylated carbenoid with
2H
-azirine.
3. Formal [2+2+2]-Cycloaddition
An approach towards phosphoryl-substituted pyridines, which is probably one of the most versatile, is based on the formal [2+2+2]-cycloaddition. Tanaka’s research group [9][10][71,72] described synthesis of annulated pyridine-2-phosphonates 26 based on rhodium(I)/biaryl-bisphosphine-complex-catalyzed cycloaddition of 1,6- and 1,7-diynes 24 with diethyl phosphorocyanidate (25) (Scheme 5). The reaction has a broad scope with respect to diynes since quaternary carbon-, methylene-, nitrogen-, and oxygen-linked internal 1,6-diynes and terminal biaryl-linked 1,7-diynes can be involved in the heterocyclization. Steric and electronic variations in diynes had minimal impact on the efficacy of the reaction, but, in some cases, using unsymmetrically substituted diynes, the reactions afforded mixtures of regioisomers. The authors proposed rhodium cyclopentadiene 27 or rhodium azacyclopentadiene 28 as two possible key intermediates in the reaction (Scheme 5). Ring expansion of both cyclopentadienes can provide seven-membered intermediate 29, and reductive elimination of Rh(I)+ from the latter accomplishes formation of the final product 26. Based on the outcome of the enantioselective version of the reaction, the authors were inclined to believe that the main reaction pathway involves intermediate 28.
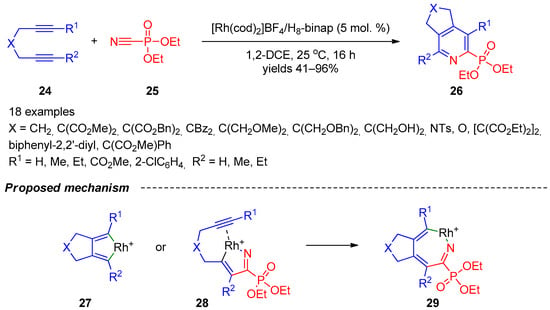
Scheme 5.
Formal [2+2+2]-cycloaddition of 1,6- and 1,7-diynes with diethyl phosphorocyanidate.
4. Phosphorylation of Pyridines
Phosphorylation is definitely the most general route to phosphorus-substituted pyridines. Due to a wide range of available phosphorylating agents and the possibility of performing the reaction in a nucleophilic, electrophilic, or radical manner, phosphorylation has attracted great attention. Such methods as the Arbuzov reaction, Hirao coupling, palladium-catalyzed cross-coupling of diethyl phosphonate with halogen-substituted heterocycles, and C(sp2)H-phosphorylation of heterocycles with diethyl phosphonates promoted by one-electron oxidants were extensively developed in past decades. Their specific applications in recent years are discussed in more detail later in the text.
4.1. Radical Phosphorylation of Pyridines
P-centered radicals can easily be generated through hydrogen atom transfer or single-electron transfer using peroxides, metal salts, and photocatalysts. Thus, radical phosphorylation has become an efficient strategy for synthesis of structurally diverse phosphorus(V)-substituted pyridines. Due to environmental friendliness and potential industrial application, photocatalytic radical phosphorylation of the pyridine ring received extra attention. In 2018, Yuan et al. [11][73] reported synthesis of 2- and 3-phosphine oxide-substituted pyridines 31 and 33 (Scheme 6, lines a,b) from 2- and 3-halopyridines 30 and 32 by photocatalytic functionalization with secondary phosphine oxides in the presence of tBuOK. The reaction occurred under mild conditions using irradiation with a blue-light-emitting diode. The scope of this transformation is somewhat limited to pyridines with electron-donating substituents and their analogs with an extended π-system. The plausible reaction mechanism involves formation of a complex of halopyridine with potassium tert-butoxide 34, absorption of a light quantum, transition to an excited state 35, and electron transfer from tert-butoxide to a pyridine ring to form the tert-butoxy radical and halopyridine radical anion 37. Elimination of halide from radical anion 36 affords aryl radical 37. Simultaneously, the tert-butoxy radical causes the proton abstraction from secondary phosphine to form a phosphorus-containing radical. Recombination of the latter radical with an aryl radical affords the final product. Recently, the approach proposed by Yuan et al. was expanded [12][13][74,75], including electron-primed photoredox [14][76] and visible light-induced nickel-catalyzed photoredox [15][77] conditions for radical generation.
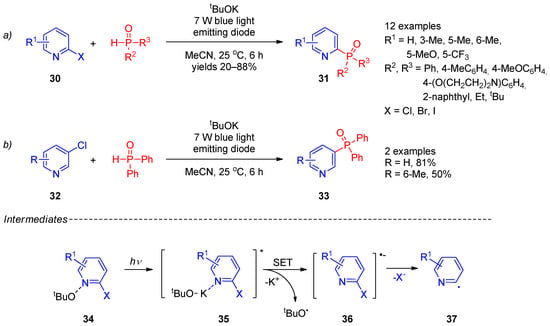
Scheme 6. Photocatalytic phosphorylation of halopyridines: (a) Synthesis of 2-phosphine oxide-substituted pyridines; (b) Synthesis of 3-phosphine oxide-substituted pyridines.
In 2019, the photocatalytic radical reaction of pyridylazo sulfones 38 with triphenyl phosphite giving pyridine-3-phosphonates 39 was described by Qiu et al. [16][78] (Scheme 7). The reaction occurred in the presence of water. A variety of substituted pyridines can be efficiently employed in this reaction. The proposed mechanism for this transformation involves excitation of arylazo sulfone 38 under visible light to provide radical 40, its decomposition into sulfonyl and aryl radicals, along with extrusion of a nitrogen molecule. Then, aryl radical 41 reacts with triphenyl phosphate to form phosphorus-centered radical 42, followed by its oxidation with the sulfonyl radical and elimination of phenol to provide the final product 39.
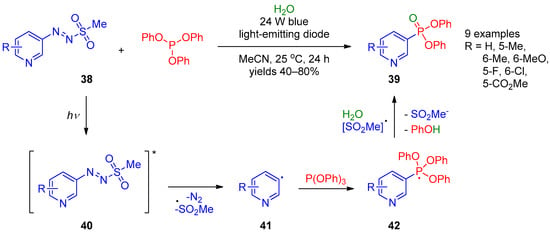
Scheme 7.
Synthesis of pyridine-3- and pyridine-4-phosphonates from pyridylazo sulfones.
Independently, in 2019, Kim et al. [17][79] described site-selective synthesis of pyridines 44 bearing phosphine oxide moieties at C-4 using radical coupling of N-ethoxypyridinium salts 43 with secondary phosphine oxides under photocatalytic conditions (Scheme 8). Further, 3-Diphenylphosphoryl-6-methoxy-1-methyl-2(1H)-quinolinone under blue-light-emitting diode illumination was used as a photocatalyst and potassium persulfate as an oxidant. Examination of the reaction scope revealed that a variety of electron-withdrawing and electron-donating substituted pyridines, as well various aryl-substituted phosphine oxides, were tolerated. The plausible mechanism of this reaction involves single-electron transfer from the photocatalyst to N-ethoxypyridinium 43, giving radical 45, which undergoes decomposition accompanied by elimination of pyridine. The remaining ethoxy radical abstracts a proton from phosphine, followed by addition of the resulting phosphinyl radical 46 to N-ethoxypyridinium 43. The subsequent deprotonation of intermediate 47 and elimination of the new ethoxy radical from intermediate 48 afford target product 44. The origin of reaction chemoselectivity was revealed by DFT calculations, showing that phosphinoyl radicals are too large for providing an electrostatic attraction between the its oxo functionality and the pyridine nitrogen crucial for ortho functionalization.
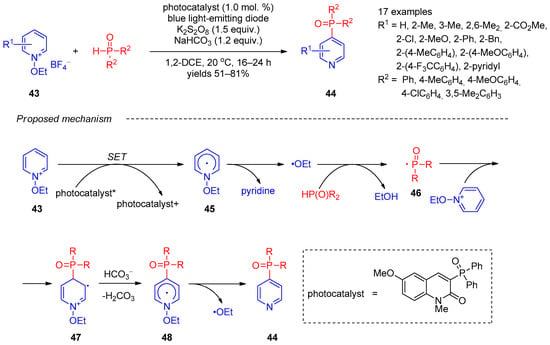
Scheme 8.
Photocatalytic reaction of
N
-ethoxypyridinium salts with phosphine oxides.
Apart from the approaches based on photocatalytic radical phosphorylation, several examples using metal salts for P-radical generation were described. Huang and co-workers [18][80] developed a CH-phosphorylation method for synthesis of pyridinyl-2-phosphonates 50 based on Ag(I)-catalyzed reaction of pyridines 49 with dialkyl phosphonates using potassium persulfate as an oxidant (Scheme 9). An interesting feature of this reaction is that it involves subsequent treatment of the reaction mixture with sodium thiosulfate, which makes it possible to significantly increase the product yield due to a reduction of pyridine N-oxide formed as a by-product. In the authors’ opinion, this transformation proceeds through a radical pathway and begins with oxidation of Ag(I) to Ag(II) with persulfate, followed by oxidation of dialkyl phosphite with Ag(II) to form radical cation 5. Addition of the latter at 2 position of the pyridine ring provides intermediate 52. Subsequent abstraction of two protons accompanied by oxidation of radical 52 affords the final product. Recently, Kittikool et al. [19][81] expanded the scope of this phosphorylation approach to 2-pyridones using Mn(OAc)2 as the catalyst.
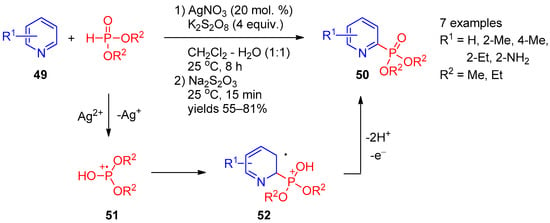
Scheme 9.
Oxidative coupling of pyridines with dialkyl phosphites in the presence of Ag(I).
Noteworthy also is a one-pot three-step protocol for synthesis of diphenyl(pyridin-2-yl)phosphine oxide based on the KOH-promoted oxidative radical phosphorylation of 2-bromopyridine with diphenylphosphine developed by Chen et al. [20][82].
4.2. Nucleophilic Phosphorylation of Pyridines
Studies by Trofimov’s research group [21][22][23][83,84,85] have contributed to recent progress in nucleophilic phosphorylation of pyridines at the 4 position. They accomplished synthesis of 4-phosphine oxide-substituted pyridines 55 by coupling of pyridines 53 with secondary phosphine oxides using diphenyl ethynyl ketone (54) as the oxidant (Scheme 10). A variety of substituted aryl and alkyl phosphine oxides can efficiently be employed in this reaction. The proposed mechanism for this transformation involves aza-Michael reaction of pyridine 53 with acetylene 54, providing intermediate 56, and deprotonation of phosphine, resulting in formation of the anion. Subsequent addition of a phosphine anion at the 4 position of the activated pyridine ring affords dihydropyridine 57. The latter undergoes isomerization to intermediate 58, followed by elimination of alkene 59 to form the final product. In this approach, 3-phenyl-2-propynenitrile can also be applied as an oxidant [22][84], The reaction can be stopped at the of step of 1,4-dihydropyridines 57 using terminal acylacetylenes [23][85]. It is interesting that 1,4-dihydropyridines are shown to be produced through 2-4 migration of the POR2 group in 1,2-dihydro adducts during vinylation/phosphorylation of pyridines [23][85]. DFT calculations supported the hypotheses that 1,4-dihydropyridines are the thermodynamic products while their 1,2-regioisomers are the kinetic ones [23][85].
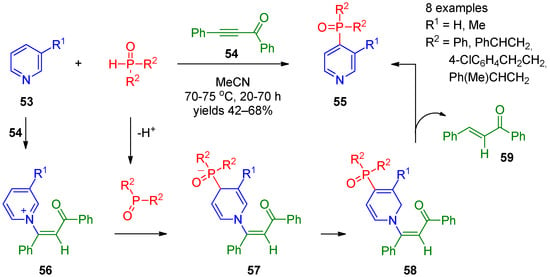
Scheme 10.
Coupling of pyridines with secondary phosphine oxides in the presence of acetylenes.
Direct nucleophilic phosphorylation at the 2 position of the pyridine ring was described in 2012 by Oka et al. [24][86] A series of 2-phosphinate-substituted pyridines 61 were prepared from N-methoxypyridinium tosylates 60 by reaction with secondary phosphinates (Scheme 11). Presumably, the reaction proceeds via the SNAr mechanism. Further, 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) in acetonitrile at low temperature was found to be the optimal base for chemoselective transformation. The procedure was general for thiophosphinate, providing 2-pyridyl thiophosphinate. The 2-pyridyl phosphinate derivatives produced by this method are P-chiral, and the reaction can be accomplished with high diastereoselectivity using chiral phosphines.
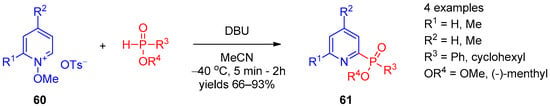
Scheme 11.
Reaction between
N
-methoxypyridinium tosylates and secondary phosphines.
In 2014, Wang et al. [25][87] showed that synthesis of pyridine-2-phosphonates can be accomplished directly from pyridine N-oxides and dimethyl phosphate over long-term heating in toluene. Independently, Lee et al. [26][88] developed an approach to synthesize diethyl pyridine-2-phosphonates 63 by reaction of pyridine N-oxides 62 with triethyl phosphite in the presence of ethyl chloroformate (Scheme 12). This reaction presumably starts with activation of pyridine N-oxide 62 by chloroformate, followed by addition of triethyl phosphate to the resulting pyridinium salt 64 to form intermediate 65. The nucleophilic attack of the chloride anion at the ethyl moiety followed by elimination of ethyl carbonate from 1,2-dihydropyridine 66 affords the final product. This approach is general for preparation of quinolin-2-ylphosphonates. An apparent limitation of this approach is formation of phosphorylated isomeric mixtures in the case of 3-bromopyridine N-oxide.
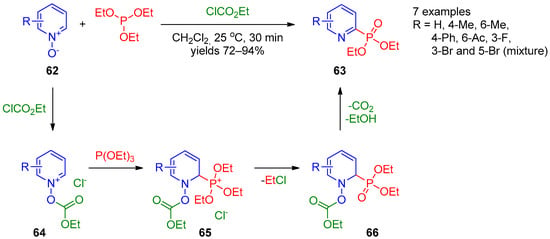
Scheme 12.
Reaction of pyridine
N
-oxides with triethyl phosphite.
Recently, Tsantrizos and co-workers [27][89] proposed a route to P-chiral 2-phosphine oxide-substituted pyridines (Scheme 13). Using the reaction of (R)-N-(1-(5-chloro-2-hydroxyphenyl)ethyl)-4-methylbenzenesulfonamide (67) with Grignard to generate chiral phosphorylating agent 68, they accomplished enantioselective synthesis of products 70 from 2-fluoropyridines 69.
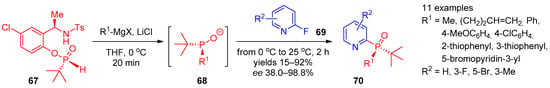
Scheme 13.
Asymmetric synthesis of tertiary pyridine-containing phosphine oxides.
4.3. Transition-Metal-Catalyzed Phosphorylation of Pyridines
Transition metals are widely used to increase efficiency of conventional non-catalyzed phosphorylation of heterocycles [28][27]. In the past decade, significant progress in the field of phosphorylation of pyridines was made due to application of palladium and nickel catalysis, which enabled expansion of the range of substrates active in phosphorylation reactions to involve halogen-, hydroxyl-, boronic acid-, triflate-, nonaflate-, ester-substituted, trimethylammonium pyridines, etc. It was also shown that transition-metal-catalyzed phosphorylation with phoshine oxides, phosphates, and phosphites could be efficiently used for construction of functionalized pyridines, including chiral structures. Meanwhile, transition-metal-catalyzed C–H phosphorylation of pyridines still remains an unsolved challenge.
Halopyridines (Hal = Cl, Br, and I) are efficient precursors for synthesis of 2,3,4-POR2-substituted pyridines under palladium catalysis conditions. In 2017, Han et al. [29][90] reported stereoselective palladium-catalyzed cross-coupling of 2-bromopyridine with chiral tert-butyl-containing phosphine oxides (Scheme 14). Use of Pd2(dba)3 with the dppp ligand enabled synthesis of tertiary pyridine-containing phosphine oxides 71 with excellent selectivity. In 2016, Dziuganowska et al. [30][13] used the same catalytic system to prepare a series of phosphonopyridinecarboxylic acid esters from appropriate bromides.
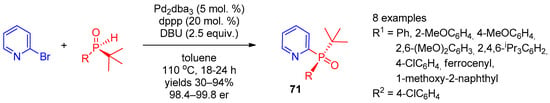
Scheme 14.
Synthesis of chiral tertiary pyridine-containing phosphine oxides.
Mykhailiuk and co-workers [31][91] expanded the scope of this reaction using the Pd2(dba)3/Xantphos catalytic system in the reaction of bromo(iodo)pyridines with dimethylphosphine oxide (Scheme 15). The scope of this reaction was found to be quite general and enables facile preparation of 2-, 3-, and 4-dimethylphosphine-oxide-substituted pyridines 72 in up to good yields. A variety of electron-withdrawing and electron-donating aryl-substituted pyridines successfully participated in the reaction.
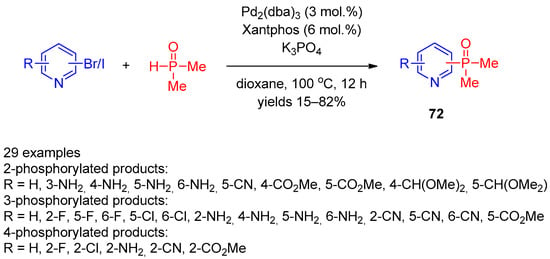
Scheme 15.
Cross-coupling of halopyridines with dimethylphosphine oxide.
Borisova and co-workers [32][92] accomplished synthesis of bis(phosphoryl)pyridines and 2,2′-bipyridines using palladium acetate/dppf-catalyzed cross-coupling of chloropyridines with secondary phosphine oxides. Ligand-free microwave-assisted Pd(OAc)2-catalyzed phosphorylation of bromopyridines with diphenylphosphine oxide or diethyl phosphite was described by Henyecz et al. [33][93]. Catalyst Pd(OAc)2 in the presence of sodium iodide as the promoter was also shown to be an effective catalyst for ligand-free coupling of diphenylphosphine oxide with pyridinium nonaflate [34][94]. An example of Pd(PPh3)4-catalyzed coupling of 2-bromo-6-pyrrolylpyridine with diethyl phosphite was recently reported by Ti et al. [35][95].
The palladium-catalyzed Hirao coupling of bromopyridines with triethyl phosphite enables preparation of pyridinephosphonates. Adam et al. [36][96] used the palladium-catalyzed cross-coupling of 3-bromopyridines 73 with triethyl phosphite to synthesize 2-aminopyridine-3-phosphonates 74 (Scheme 16). It is worth noting that the reaction was performed in the absence of the solvent during short-term heating to 160–180 °C.
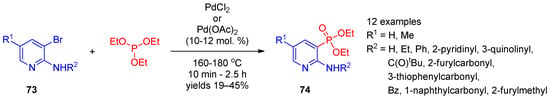
Scheme 16.
Pd(II)-catalyzed cross-coupling of 3-bromopyridines with triethyl phosphite.
Apart from halopyridines, pyridine boronic acids and hydroxypyridines were involved in phosphorylation under palladium catalysis. Zou, Wu, and co-workers [37][97] constructed a C–P bond by means of cross-coupling of pyridine boronic acids 75 with dialkyl phosphonates under PdCl2 catalysis conditions (Scheme 17). The reaction requires the presence of Ag2O as the oxidant. The method can be used to prepare structurally diverse pyridine-3- and pyridine-4-phosphonates 76.
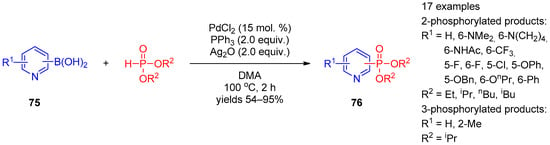
Scheme 17.
Cross-coupling of pyridine carboxylic acids with dialkyl phosphites.
Recently, Ding and co-workers [38][98] described the general palladium-catalyzed one-pot procedure for synthesis of phosphonates, phosphinates, and phosphine oxides from phenols mediated by sulfuryl fluoride. The reaction was efficient for functionalization of 2- and 3-hydroxypyridines 77 with diethyl phosphonate (Scheme 18). According to the proposed mechanism, the fluorosulfates generated in situ are key intermediates in synthesis of pyridines 78.
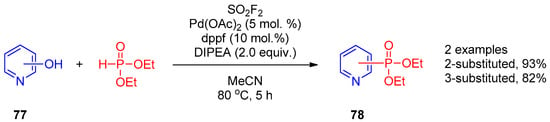
Scheme 18.
Replacement of the OH group of hydroxypyridines mediated by sulfuryl fluoride.
Before these works, in 2012, direct replacement of an OH group of hydroxypyridines by a diphenylphosphoryl moiety was achieved by Zhao et al. [39][99] under Ni(II) catalysis. A C–O bond was activated using bromotripyrrolidinophosphonium hexafluorophosphate. The activated complex of hydroxypyridine 77 with this salt reacts with diphenylphosphine oxide in the presence of dichloro[1,3-bis(diphenylphosphino)propane]nickel salts as the catalyst to produce pyridines 79 (Scheme 19).
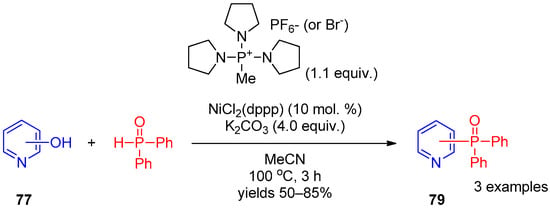
Scheme 19.
Replacement of the OH group of hydroxypyridines by the diphenylphosphoryl moiety.
Later, the substrate scope of Ni-catalyzed phosphorylation of pyridines with phosphine oxides was expanded to pyridine carboxylic acid phenyl esters [40][100] and trimethylammonium triflate of pyridine [41][101]. Cross-coupling of pyridine tosylates [42][102] and aryltrimethylammonium tetrafluoroborate [43][103] with phosphites was also implemented.
A relatively general method was proposed by Yamaguchi’s research group [40][100]. They performed cross-coupling of pyridine carboxylic acid phenyl esters 80 with secondary phosphine oxides accompanied by decarbonylation (Scheme 20). The reaction is catalyzed by nickel acetate and requires high temperatures (150–170 °C). This approach is applicable to synthesis of 2- and 3-phosphine-oxide-substituted pyridines 81, the position of the POR2 substituent in the product being defined by the position of the carbonyl group in the starting compound. The authors suggested that this transformation initially proceeds through oxidative insertion of Ni(0) into the C–O bond, providing intermediate 82, and exchange of the phenoxide ligand by phosphine provides 83. Subsequent decarbonylation of intermediate 83 and reductive elimination from intermediate 84 afford the final product.
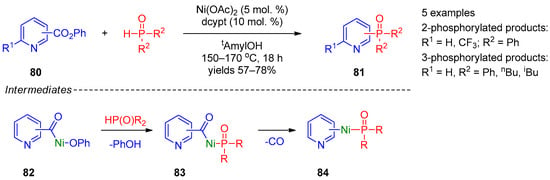
Scheme 20.
Cross-coupling of aryl carboxylic acid esters with phosphine oxides.
Of interest as well is Ni(II)-catalyzed reaction of tosylates 85 with H-phosphonate diesters proposed by Chun-Jing Li [42][102] as an approach to 2- and 3-phosphorylpyridines 86; NiCl2(cod)2 was an optimal catalyst for this transformation (Scheme 21).
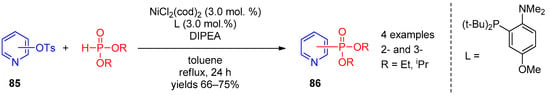
Scheme 21.
Ni(II)-catalyzed replacement of the tosyl group in pyridines.