Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Wararat Chiangjong.
The detection of genomic aberrations in cancers has yielded a wealth of information to discover oncogenic drivers or pathogenic variants that are relevant for the development of precise treatment strategies. Studies have shown promising outcomes in adult cancer patients with well characterized cancer genetic biomarkers.
- precision medicine
- pediatric solid tumor
- actionable mutations
1. Introduction
Cancer occurrence before the age of 20 years is rare, but it is one of the leading causes of disease-related mortality in children and adolescents globally [1,2][1][2]. Approximately 300,000 children aged 0–19 years old worldwide are diagnosed with cancer each year [1], and 80% of these patients live in low- and middle-income countries (LMCs). Hematologic malignancies are more common among pediatric cancers, comprising about half of all cases. Solid malignancies are rarer and heterogenous as following an age-specific pattern. In early childhood, embryonal-type solid tumors are common, such as neuroblastoma, retinoblastoma, medulloblastoma, hepatoblastoma, and Wilms tumor [3]. The prognosis for childhood cancer has improved dramatically over the past four decades, particularly for hematologic malignancies [2]. Nonetheless, treatment outcomes for childhood solid malignancies remain unsatisfactory, especially in LMCs [4,5][4][5].
Genetic sequencing studies have led to the identification of somatic gene alterations as cancer hallmarks and germline predisposition and targeted the molecular abnormalities for the development of precise treatment [6,7,8][6][7][8]. Dramatic differences in the genetic repertoire between normal and cancer cells provide advantages of molecular targeted therapies over traditional strategies based on the target selectivity [9,10,11][9][10][11]. Several components in cellular signaling pathways, i.e., tyrosine receptor kinase (TRK), mitogen-activating protein kinase (MAPK) and phosphoinositide 3-kinases (PI3K)-mammalian target of rapamycin (mTOR), have been commonly identified as actionable mutations that would recommend appropriately targeted therapies [12,13][12][13]. These generic biomarker-driven precise treatments have been investigated in several pre-clinical and clinical trials since the early 2000s [14].
Progress in designing treatments targeting molecular alterations specific to pediatric cancers is considerably slow due to the rare and unique genetic alterations in children compared to adults [15]. A report from the European Union (E.U.) revealed that up to 26 anticancer drugs approved for adults might be also effective in pediatric malignancies; however, only four of these drugs have been approved for childhood cancers [16]. Nishiwaki S. and Ando Y. reported that only 3 out of 66 drugs with adult indications have been approved for pediatrics in the E.U., United States, and Japan [17]. Thus far, larotrectinib and entrectinib have been two of the most successful molecularly targeted therapies for children with solid tumors and have shown their promising responses in patients with NTRK-fusion [9]. In 2018, larotrectinib became the first drug to receive FDA approval to treat NTRK fusion-positive solid tumors in children and adults [18]. Similarly, entrectinib, a multi-kinase inhibitor, also received approval for the treatment of TRK fusion solid tumors in patients aged ≥ 12 years [19].
2. Genetic Alterations on Cancer Hallmarks
2.1. Cancer Hallmarks and Common Targeted Signaling Pathways
Cancers are driven by changes in cellular DNA which further promote the transition of genetic landscape, especially in cell survival programs, leading to unstoppable cell growth with abnormal cellular characteristics [20]. In contrast to normal tissues, cancer cells can dysregulate their own signaling cascades autonomously, thus controlling their own cell fate [21]. Besides their proficiency in cancer hallmarks in evading growth suppressors, resisting cell death, reprogramming cellular mechanisms, and avoiding immune destruction, cancer cells can also acquire the capability to sustain proliferative signaling in several alternative ways [22,23][22][23]. Cancer cells may send signals to activate normal cells within the tumor parenchyma, which reciprocally communicate to supply cancer cells with various growth-promoting factors [24,25][24][25]. Furthermore, common downstream components in distinct signaling cascades also allowed cancer cells to control cell fate in a growth factor-independent manner by triggering the downstream molecules directly, negating the need for ligand-mediated receptor activation [23,26][23][26]. Hence, the vast majority of different cancers are coordinately modulated by canonical oncogenic drivers, including KRAS, MYC, NOTCH, and TP53. This factors highlights the need to fully elucidate their regulatory networks for further therapeutic development [27].2.2. Tumor Cells Have Both Germline and Somatic Variants in Their Genome
Cancer gene mutations can be either inherited or acquired. Hereditary or germline mutations refer to the genomic changes that occur in germ cells and can be detected in all cells of the offspring and are passed inter-generationally [28,29][28][29]. Genetic predisposition has been described by certain characteristics, including [30];-
Familial history of the same or related cancers;
-
Occurrence of bilateral or multifocal cancers;
-
Earlier age at disease onset;
-
Physical suggestive of a predisposition syndrome;
-
Appearance of specific tumor types corresponding to the genetic predisposition.
2.3. Germline and Somatic Variants Classified as Druggable
In the context of defining mutational actionability, the relevant effects of genomic aberration participating in cancer phenotypes are considered. DNA aberrations include missense, nonsense, frameshift mutations, and chromosome rearrangements, with some changes affecting only a single DNA base that may or may not alter the protein’s property and some point mutations completely abrogating protein expression. A wide variety of gene alterations have been detected such as activating point mutation in BRAF, ALK, EGFR and FGFR1 genes, high copy number gains in PDGFRA and ERBB2, loss-of-function mutation affecting PTEN, PTPN11, PIK3R1, and MTORC1, CDKN2A/2B deletions, or in-frame expression of large indels (NOTCH1 and FOXA1) [12]. Other changes involving larger stretches of DNA may include rearrangements, deletions, or duplications of long stretches of DNA [54][39]. For example, exon skipping on MET exon 14 proto-oncogenes resulting from intronic mutation increases the protein lifespan and promotes MET activation in lung carcinogenesis [55][40]. The significance of genetic variants may vary depending upon their potential effects on cellular functions. An “actionable” mutation is defined as a genetic aberration that is potentially responsive to targeted therapy, while a “driver” mutation refers to variants that confer a growth advantage to cancer cells but may not be targetable with a specific treatment yet. Passenger mutation is used to designate cancer-neutral variations and is unlikely to be under selective pressure during the evolution of the cancerous cells [56,57][41][42]. The “passenger” mutation has the lowest tendency to impact protein function, most of which are synonymous substitutions; however, these mutations occur more frequently than driver or actionable mutations. Unraveling the passenger mutational paradigm has otherwise revealed the existence of pre-existing latent driver mutations in which certain combinations of the passenger mutations could indeed be functional drivers. One example is the non-hotspot, passenger mutation of the Akt1 gene at position L52R, C77F, and Q79K, which promotes its membrane localization similarly to the E17K driver. In contrast, the co-existence of D32Y, K39N, and P42T passenger mutations can lead to Akt conformational inactivation, suggesting that treatment decisions based only on genetics may overlook crucial actionable components [56,58][41][43]. In addition, silent mutations occurring near the donor splice junction could contrarily affect exon splicing. For example, T125T mutation in TP53 is a recurrent mutation that is generally considered a non-functional passenger event; however, its existence at the −1 donor site of exon 4 raises the possibility that this mutation affects splicing. Further integration with RNA-seq data demonstrated that T125T mutation resulted in the retention of intron 4 and introduced a premature stop codon such as nonsense-mediated decay [59][44]. Thus, aberrant splicing caused by silent mutations should be carefully evaluated during interpretation of the sequencing results. The accumulated data of genetic composition data from the tumors of patients has become a growing compendium of molecular biomarkers for precise treatment with FDA-approved drugs. Figure 1 summarizes the actionable mutations currently approved by FDA consortium for targeted therapy in adult cancers and pediatric solid tumors.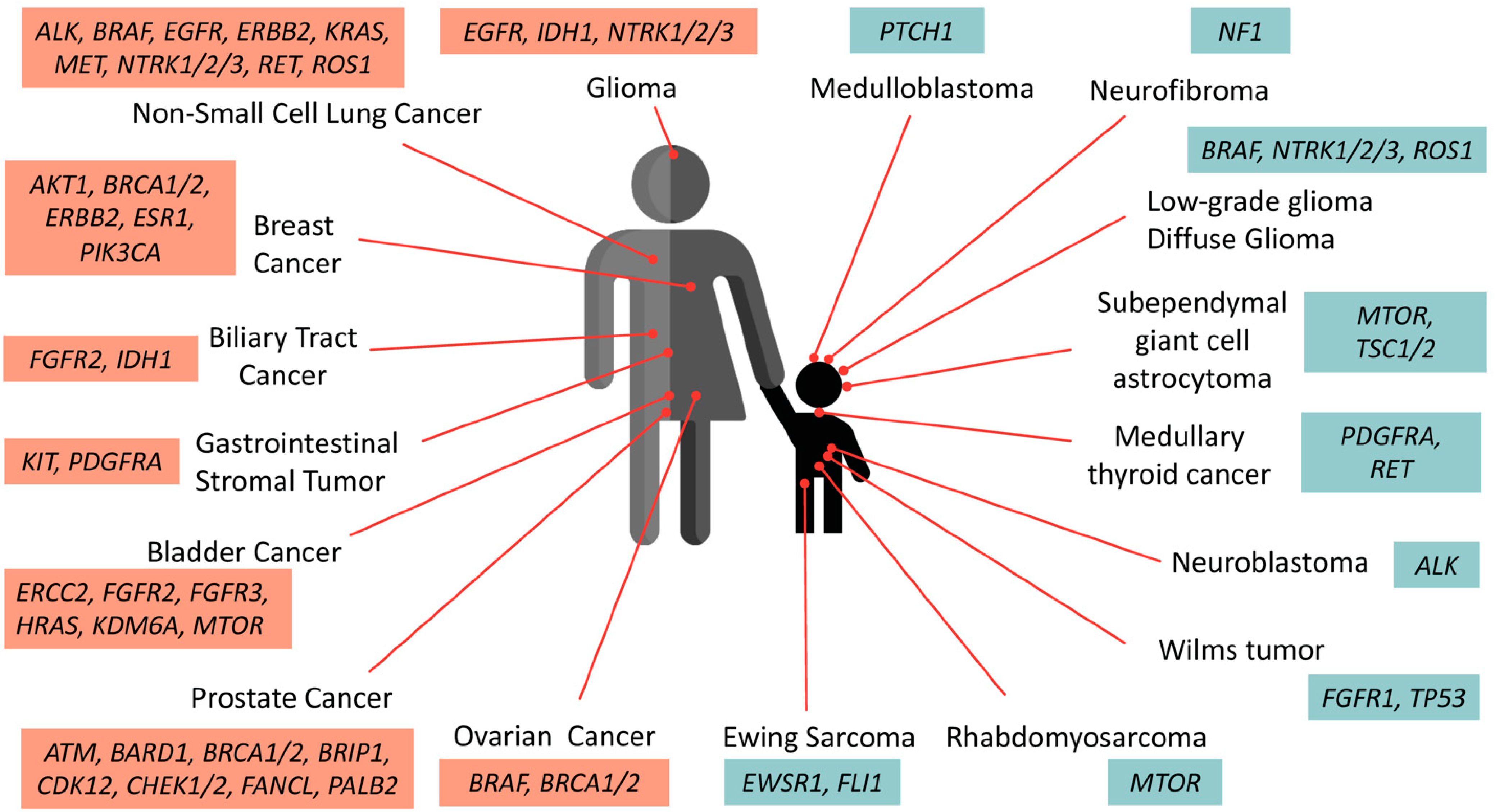
Figure 1. Oncogenic drivers identified in adult and pediatric solid tumors. These selective biomarkers are predicted to be responsive to various levels of FDA-approved drugs. Note that targeted therapies against PTCH1 and ALK in medulloblastoma and neuroblastoma are currently undergoing clinical assessment and awaiting further approval.
3. Pediatric Cancer Genome
3.1. Pediatric vs. Adult Cancer Development
Pediatric cancers reflect a heterogeneous group of disorders distinct from adult cancers in terms of cellular origins, genetic complexity, and specific driver alterations [62,63][45][46]. Pediatric malignancies typically occur in developing mesoderm rather than adult epithelia (ectoderm) and are often induced by inherited or sporadic errors during development [33]. Studies have quantified the mutation burden in many pediatric cancers, identifying approximately 5 to 10 protein-coding variants identified across multiple tumor types except in osteosarcoma, which showed an average of 25 protein-affecting mutations. In contrast, the average number of mutations in adult cancers ranges between 33 to 66 in pancreatic, colon, breast, and brain cancers while mutagen-caused adult tumors (such as melanoma and lung cancers) can include up to 200 protein-coding variants [64,65,66][47][48][49]. At diagnosis, patients with pediatric cancers tend to have less complexity on mutational spectra than those in adult cancers; however, with treatment-refractory tumors and recurrence—the mutation rates in pediatric tumors have increased to be comparable to adult tumors [67,68][50][51]. Moreover, the rare occurrence of pediatric cancers and the low frequency of recurrent genomic alterations have a great impact on the investigations and the availability of targeted agents. Thus, there is an urgent need to accelerate the pace of genomic data acquisition and clinical trials in children to design more effective strategies for pediatric precision oncology.3.2. Somatic and Germline Mutations Identified in Pediatric Cancer Cohorts
Single nucleotide variations (SNVs) and small indels are the usual mutations identified in adult cancers. In contrast, childhood cancers show a relatively high prevalence of copy number aberrations (CNAs) and specific structural variations (SVs). Note that insertion and deletion lead to adding and removing at least one nucleotide to the gene, respectively, which can affect protein functions and contribute to carcinogenesis. Current data suggest that approximately 10% of pediatric cancers are caused by genetic predisposition [32]. Zhang et al. [31] revealed that 95 out of 1120 (8.5%) patients younger than 20 years of age harbor germline mutations in cancer-predisposing genes. Diets et al. [69][52] performed trio-based whole-exome sequencing on the germline DNA of 40 selected children with cancer and their parents. Of these, germline pathogenic mutations were identified in 20% (8/40) of children with cancer [69][52]. Similarly, Grobner et al. [33] reported that most germline variants were related to DNA repair genes from mismatch (MSH2, MSH6, PMS2) and double-stranded break (TP53, BRCA2, CHEK2) repair. Using combined somatic and germline sequencing for children with solid tumors, Parsons et al. [32] identified actionable mutations in up to 40% (47/121) of pediatric solid tumor tissues. Likewise, Wong et al. [12] performed the combination of tumor and germline sequencing (WGS) and RNA sequencing (RNA-seq) to identify 968 reportable molecular aberrations (39.9% in both WGS and RNA-seq; 35.1% in WGS only and 25.0% in RNA-seq only) in 247 high-risk pediatric cancer patients with 252 tumor tissues. Interestingly, 93.7% of these patients had at least one germline or somatic aberration, 71.4% had therapeutic targets, and 5.2% had a change in diagnosis [12]. These cohort studies emphasized that comprehensive molecular profiling could resolve molecular aberration in high-risk pediatric cancer and provide clinical benefits in a significant number of patients. In the era of next-generation sequencing, publicly genomic data access is considered one of the keys to accelerate research. The St. Jude Cloud is one of the most promising data-sharing ecosystems, with genomic data from >10,000 pediatric patients with cancer and long-term survivors. When exploring the mutational profile of pediatric solid tumors, the resource has revealed common genetic alterations among the different cancer types. This integrative view of genomic data could be further used to expedite studies of pediatric cancer-associated risk factors and initiate novel therapeutic investigations for improving treatment outcomes.4. Current Progress in Clinical Trials for Pediatric Precision Oncology
Genomic precision medicine has demonstrated preferential outcomes among ongoing genomic-driven clinical trials in adult cancers. Yet, clinical investigations based on pediatric tumor genetics are still lacking. Based on the patient genetic profile screening, scattered reports on molecularly defined pediatric patients are showing prominent responses to some targeted therapies. For example, targeting ALK has shown success in treatments of ALK(+) non-small cell lung cancers and also in childhood anaplastic large cell lymphoma (ALCL) and inflammatory myofibroblastic tumor using the ALK inhibitor crizotinib [92][53]. While ALK mutation is the most common somatic mutation in neuroblastoma, crizotinib was compromised due to the interference by common ALK mutation F1174 [93][54]. Since then, ceritinib, alectinib, brigatinib, and lorlatinib have been approved against advanced ALK+ NSCLC [94,95,96,97][55][56][57][58]. Intriguingly, the third-generation TKI that targets both ALK and ROS1, lorlatinib, has recently shown promise in patients with ALK mutated neuroblastoma, but most of the studies are still at phase I clinical trial. [98][59]. Nonetheless, repotrectinib, a next-generation ROS1/TRK inhibitor with >90-fold potency against ROS1 than crizotinib in NSCLC patients is also being tested for dose escalation in phase II clinical trial with patients aged ≥ 12 years [99][60]. Another promising example is the targeted therapy against Ras-Raf-MEK-ERK signaling cascade which include somatic BRAF alterations (BRAF V600E and BRAF fusions). The prototype for targeting BRAF V600E/K is cutaneous melanoma, where 40–60% of patients with these mutations are eligible for the FDA-approved BRAF-inhibitor, vemurafenib [100][61]. Low-grade-gliomas have been identified to contain multiple alterations in Ras-Raf-MEK-ERK pathway, and a single treatment of vemurafenib in malignant glioma resulted in tumor regression [85,101][62][63]. Recently, Jain et al. [102][64] reported that a combination of BRAF-inhibitor dabrafenib and MEK-inhibitor trametinib enhanced treatment efficacies in pediatric low-grade-glioma carrying KIAA1549-BRAF fusion. Additionally, several studies have utilized the combination of molecularly targeted agents and traditional chemotherapy or radiation to reduce the severe side effects caused by an intensive dose of chemo/radiotherapy while minimizing acquired drug resistance due to selective pressure. The following large-scale pediatric and young-adult precision oncology programs have been launched with multiple-arm trials for patients with matched molecular profiles: TAPUR (ClinicalTrials.gov identifier NCT02693535), NCI-COG Pediatric MATCH (NCT03155620), the Tumor-Agnostic Precision Immuno-Oncology and Somatic Targeting Rational for You (TAPISTRY) (NCT04589845). These global, multicenter, open-label, multi-cohort studies are now at phase II, and the treatment assignment has relied on the basis of relevant onco-genotypes as identified by a Clinical Laboratory Improvement Amendments (CLIA)-certified or a validated next-generation sequencing (NGS) assay. While the eligible criteria of TAPUR are open for patients aged 12 years old or older, most of the patients enrolled are reported to have adult cancer phenotypes [103,104,105][65][66][67]. In contrast, the NCI-COG Pediatric MATCH aims to evaluate the molecular-targeted therapies with selected biomarkers of childhood and young adult patients with a reported detection rate of actionable alterations of 31.5% from the first 1000 tumors screened. Assignments to treatment arms were made for 28% of patients screened and 13% of patients enrolled in the treatment trial [106][68]. In the TAPISTRY study, nine targeted treatments are being examined, and eleven non-randomized treatment arms are available for participants of all ages with locally advanced/metastatic solid tumors. The purpose of this study is to evaluate the safety and efficacy of different targeted therapies and immunotherapies in patients as single agents, but the results of the study are still to be released. Overall, the advancements in high-throughput sequencing technology have closed the gap between the current treatment paradigm and precision medicine, markedly improving rates of response, progression-free survival (PFS), and overall survival (OS) compared to traditional randomized trials. Moreover, the multicenter, open-label, multi-arm treatment designs can further benefit treatment strategies by yielding efficacy and toxicity data in a timely manner with cost-effectiveness. Therefore, in the future, international coordination will be crucial to generate a database to inform rational trial design and to evaluate the combination of treatments/interventions that ensure more favorable outcomes.References
- Steliarova-Foucher, E.; Colombet, M.; Ries, L.A.G.; Moreno, F.; Dolya, A.; Bray, F.; Hesseling, P.; Shin, H.Y.; Stiller, C.A.; IICC-3 Contributors. International incidence of childhood cancer, 2001–10: A population-based registry study. Lancet Oncol. 2017, 18, 719–731.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30.
- Tsui, P.C.; Lee, Y.-F.; Liu, Z.W.Y.; Ip, L.R.H.; Piao, W.; Chiang, A.K.S.; Lui, V.W.Y. An update on genomic-guided therapies for pediatric solid tumors. Future Oncol. 2017, 13, 1345–1358.
- Friedrich, P.; Ortiz, R.; Fuentes, S.; Gamboa, Y.; Ah Chu-Sanchez, M.S.; Arambú, I.C.; Montero, M.; Báez, F.; Rodríguez-Galindo, C.; Antillón-Klussmann, F.; et al. Barriers to effective treatment of pediatric solid tumors in middle-income countries: Can we make sense of the spectrum of nonbiologic factors that influence outcomes? Cancer 2014, 120, 112–125.
- Lupo, P.J.; Spector, L.G. Cancer Progress and Priorities: Childhood Cancer. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1081–1094.
- Langenberg, K.P.S.; Looze, E.J.; Molenaar, J.J. The Landscape of Pediatric Precision Oncology: Program Design, Actionable Alterations, and Clinical Trial Development. Cancers 2021, 13, 4324.
- Ahmed, A.A.; Vundamati, D.S.; Farooqi, M.S.; Guest, E. Precision Medicine in Pediatric Cancer: Current Applications and Future Prospects. High-Throughput 2018, 7, 39.
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision Oncology: Who, How, What, When, and When Not? Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 160–169.
- Butler, E.; Ludwig, K.; Pacenta, H.L.; Klesse, L.J.; Watt, T.C.; Laetsch, T.W. Recent progress in the treatment of cancer in children. CA A Cancer J. Clin. 2021, 71, 315–332.
- Kumar-Sinha, C.; Chinnaiyan, A.M. Precision oncology in the age of integrative genomics. Nat. Biotechnol. 2018, 36, 46–60.
- Abrams, J.; Conley, B.; Mooney, M.; Zwiebel, J.; Chen, A.; Welch, J.J.; Takebe, N.; Malik, S.; McShane, L.; Korn, E.; et al. National Cancer Institute’s Precision Medicine Initiatives for the New National Clinical Trials Network. Am. Soc. Clin. Oncol. Educ. Book 2014, 34, 71–76.
- Wong, M.; Mayoh, C.; Lau, L.M.S.; Khuong-Quang, D.A.; Pinese, M.; Kumar, A.; Barahona, P.; Wilkie, E.E.; Sullivan, P.; Bowen-James, R.; et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med. 2020, 26, 1742–1753.
- Worst, B.C.; Van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101.
- Lander, E.S. Initial impact of the sequencing of the human genome. Nature 2011, 470, 187–197.
- DuBois, S.A.-O.X.; Corson, L.A.-O.; Stegmaier, K.; Janeway, K.A.-O. Ushering in the next generation of precision trials for pediatric cancer. Science 2019, 15, 1175–1181.
- Vassal, G.; Geoerger, B.; Morland, B. Is the European Pediatric Medicine Regulation Working for Children and Adolescents with Cancer? Clin. Cancer Res. 2013, 19, 1315–1325.
- Nishiwaki, S.; Ando, Y. Gap between pediatric and adult approvals of molecular targeted drugs. Sci. Rep. 2020, 10, 17145.
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib inTRKFusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739.
- Robinson, G.W.; Gajjar, A.J.; Gauvain, K.M.; Basu, E.M.; Macy, M.E.; Maese, L.D.; Sabnis, A.J.; Foster, J.H.; Shusterman, S.; Yoon, J.; et al. Phase 1/1B trial to assess the activity of entrectinib in children and adolescents with recurrent or refractory solid tumors including central nervous system (CNS) tumors. J. Clin. Oncol. 2019, 37, 10009.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558.
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337.
- Chiangjong, W.; Chutipongtanate, S. EV-out or EV-in: Tackling cell-to-cell communication within the tumor microenvironment to enhance anti-tumor efficacy using extracellular vesicle-based therapeutic strategies. OpenNano 2022, 8, 100085.
- Csermely, P.; Korcsmáros, T.; Nussinov, R. Intracellular and intercellular signaling networks in cancer initiation, development and precision anti-cancer therapy. Semin. Cell Dev. Biol. 2016, 58, 55–59.
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Dancey, J.E.; Bedard, P.L.; Onetto, N.; Hudson, T.J. The Genetic Basis for Cancer Treatment Decisions. Cell 2012, 148, 409–420.
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104.
- Jongmans, M.C.J.; Loeffen, J.L.C.M.; Waanders, E.; Hoogerbrugge, P.M.; Ligtenberg, M.J.L.; Kuiper, R.P.; Hoogerbrugge, N. Recognition of genetic predisposition in pediatric cancer patients: An easy-to-use selection tool. Eur. J. Med. Genet. 2016, 59, 116–125.
- Zhang, J.; Walsh, M.F.; Wu, G.; Edmonson, M.N.; Gruber, T.A.; Easton, J.; Hedges, D.; Ma, X.; Zhou, X.; Yergeau, D.A.; et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. N. Engl. J. Med. 2015, 373, 2336–2346.
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children with Solid Tumors. JAMA Oncol. 2016, 2, 616–624.
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327.
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.N.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271.
- Brodeur, G.M.; Nichols, K.E.; Plon, S.E.; Schiffman, J.D.; Malkin, D. Pediatric Cancer Predisposition and Surveillance: An Overview, and a Tribute to Alfred G. Knudson Jr. Clin. Cancer Res. 2017, 23, e1–e5.
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724.
- Murtaza, M.; Dawson, S.-J.; Tsui, D.W.Y.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112.
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376.
- Minati, R.; Perreault, C.; Thibault, P. A Roadmap Toward the Definition of Actionable Tumor-Specific Antigens. Front. Immunol. 2020, 11, 583287.
- Onozato, R.; Kosaka, T.; Kuwano, H.; Sekido, Y.; Yatabe, Y.; Mitsudomi, T. Activation of MET by Gene Amplification or by Splice Mutations Deleting the Juxtamembrane Domain in Primary Resected Lung Cancers. J. Thorac. Oncol. 2009, 4, 5–11.
- Nussinov, R.; Jang, H.; Tsai, C.-J.; Cheng, F. Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers. PLoS Comput. Biol. 2019, 15, e1006658.
- Carr, T.H.; McEwen, R.; Dougherty, B.; Johnson, J.H.; Dry, J.R.; Lai, Z.; Ghazoui, Z.; Laing, N.M.; Hodgson, D.R.; Cruzalegui, F.; et al. Defining actionable mutations for oncology therapeutic development. Nat. Rev. Cancer 2016, 16, 319–329.
- Yi, K.H.; Axtmayer, J.; Gustin, J.P.; Rajpurohit, A.; Lauring, J. Functional analysis of non-hotspot AKT1 mutants found in human breast cancers identifies novel driver mutations: Implications for personalized medicine. Oncotarget 2013, 4, 29–34.
- Zhou, X.; Edmonson, M.N.; Wilkinson, M.R.; Patel, A.; Wu, G.; Liu, Y.; Li, Y.; Zhang, Z.; Rusch, M.C.; Parker, M.; et al. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat. Genet. 2016, 48, 4–6.
- Ferrari, A.; Casanova, M.; Massimino, M.; Sultan, I. Peculiar features and tailored management of adult cancers occurring in pediatric age. Expert Rev. Anticancer Ther. 2010, 10, 1837–1851.
- Jones, D.T.W.; Banito, A.; Grünewald, T.G.P.; Haber, M.; Jäger, N.; Kool, M.; Milde, T.; Molenaar, J.J.; Nabbi, A.; Pugh, T.J.; et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat. Rev. Cancer 2019, 19, 420–438.
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Rep. 2014, 7, 104–112.
- Rahal, Z.; Abdulhai, F.; Kadara, H.; Saab, R. Genomics of adult and pediatric solid tumors. Am. J. Cancer Res. 2018, 8, 1356–1386.
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218.
- Schramm, A.; Köster, J.; Assenov, Y.; Althoff, K.; Peifer, M.; Mahlow, E.; Odersky, A.; Beisser, D.; Ernst, C.; Henssen, A.G.; et al. Mutational dynamics between primary and relapse neuroblastomas. Nat. Genet. 2015, 47, 872–877.
- Morrissy, A.S.; Garzia, L.; Shih, D.J.H.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.G.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357.
- Diets, I.J.; Waanders, E.; Ligtenberg, M.J.; van Bladel, D.A.G.; Kamping, E.J.; Hoogerbrugge, P.M.; Hopman, S.; Olderode-Berends, M.J.; Gerkes, E.H.; Koolen, D.A.; et al. High Yield of Pathogenic Germline Mutations Causative or Likely Causative of the Cancer Phenotype in Selected Children with Cancer. Clin. Cancer Res. 2018, 24, 1594–1603.
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK with Crizotinib in Pediatric Anaplastic Large Cell Lymphoma and Inflammatory Myofibroblastic Tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221.
- Bresler Scott, C.; Wood Andrew, C.; Haglund Elizabeth, A.; Courtright, J.; Belcastro Lili, T.; Plegaria Jefferson, S.; Cole, K.; Toporovskaya, Y.; Zhao, H.; Carpenter Erica, L.; et al. Differential Inhibitor Sensitivity of Anaplastic Lymphoma Kinase Variants Found in Neuroblastoma. Sci. Transl. Med. 2011, 3, 108ra114.
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.-W.; Ou, S.-H.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838.
- Soria, J.-C.; Tan, D.S.W.; Chiari, R.; Wu, Y.-L.; Paz-Ares, L.; Wolf, J.; Geater, S.L.; Orlov, S.; Cortinovis, D.; Yu, C.-J.; et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): A randomised, open-label, phase 3 study. Lancet 2017, 389, 917–929.
- Camidge, D.R.; Kim, H.R.; Ahn, M.-J.; Yang, J.C.H.; Han, J.-Y.; Hochmair, M.J.; Lee, K.H.; Delmonte, A.; García Campelo, M.R.; Kim, D.-W.; et al. Brigatinib Versus Crizotinib in Advanced ALK Inhibitor–Naive ALK-Positive Non–Small Cell Lung Cancer: Second Interim Analysis of the Phase III ALTA-1L Trial. J. Clin. Oncol. 2020, 38, 3592–3603.
- Shaw, A.T.; Bauer, T.M.; De Marinis, F.; Felip, E.; Goto, Y.; Liu, G.; Mazieres, J.; Kim, D.-W.; Mok, T.; Polli, A.; et al. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029.
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016, 6, 96–107.
- Drilon, A.; Ou, S.-H.I.; Cho, B.C.; Kim, D.-W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.-J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) Is a Next-Generation ROS1/TRK/ALK Inhibitor That Potently Inhibits ROS1/TRK/ALK Solvent—Front Mutations. Cancer Discov. 2018, 8, 1227–1236.
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 809–819.
- Del Bufalo, F.; Carai, A.; Figà-Talamanca, L.; Pettorini, B.; Mallucci, C.; Giangaspero, F.; Antonelli, M.; Badiali, M.; Moi, L.; Bianco, G.; et al. Response of recurrent BRAFV600E mutated ganglioglioma to Vemurafenib as single agent. J. Transl. Med. 2014, 12, 356.
- Robinson, G.W.; Orr, B.A.; Gajjar, A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 2014, 14, 258.
- Jain, P.; Silva, A.; Han, H.J.; Lang, S.-S.; Zhu, Y.; Boucher, K.; Smith, T.E.; Vakil, A.; Diviney, P.; Choudhari, N.; et al. Overcoming resistance to single-agent therapy for oncogenic BRAF gene fusions via combinatorial targeting of MAPK and PI3K/mTOR signaling pathways. Oncotarget 2017, 8, 84697–84713.
- Al Baghdadi, T.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Rich, P.; Ahn, E.R.; Chai, S.; Rygiel, A.L.; Osayameh, O.; Antonelli, K.R.; et al. Sunitinib in Patients with Metastatic Colorectal Cancer (mCRC) with FLT-3 Amplification: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Target. Oncol. 2020, 15, 743–750.
- Fisher, J.G.; Tait, D.; Garrett-Mayer, E.; Halabi, S.; Mangat, P.K.; Schink, J.C.; Alvarez, R.H.; Veljovich, D.; Cannon, T.L.; Crilley, P.A.; et al. Cetuximab in Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Ovarian Cancer without KRAS, NRAS, or BRAF Mutations: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Target. Oncol. 2020, 15, 733–741.
- Alva, A.S.; Mangat, P.K.; Garrett-Mayer, E.; Halabi, S.; Hansra, D.; Calfa, C.J.; Khalil, M.F.; Ahn, E.R.; Cannon, T.L.; Crilley, P.; et al. Pembrolizumab in Patients with Metastatic Breast Cancer with High Tumor Mutational Burden: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. J. Clin. Oncol. 2021, 39, 2443–2451.
- Parsons, D.W.; Janeway, K.A.; Patton, D.R.; Winter, C.L.; Coffey, B.; Williams, P.M.; Roy-Chowdhuri, S.; Tsongalis, G.J.; Routbort, M.; Ramirez, N.C.; et al. Actionable Tumor Alterations and Treatment Protocol Enrollment of Pediatric and Young Adult Patients with Refractory Cancers in the National Cancer Institute–Children’s Oncology Group Pediatric MATCH Trial. J. Clin. Oncol. 2022, 40, 2224–2234.
More