Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Saleh Jamehdor.
The most widely used genome editing toolkit is CRISPR (clustered regularly interspaced short palindromic repeats). It provides the possibility of replacing and modifying DNA and RNA nucleotides. Furthermore, with advancements in biological technology, inhibition and activation of the transcription of specific gene(s) has become possible. Bioinformatics tools that target the evolution of CRISPR-associated protein 9 (Cas9) turn this protein into a vehicle that is specific for a DNA or RNA region with single guide RNA (sgRNA). This toolkit could be used by researchers to investigate the function of stem cell gene(s).
- CRISPR
- genome editing
- stem cells
- Gene therapy
- DNA and RNA manupulation
1. Introduction
Stem cells are heterogeneous and unspecialized cells that are the foundation of every organ and cell in our body [1]. Depending on their origin, stem cells are categorized into embryonic stem cells (ESCs), which exist in the inner cell mass (ICM) at an early stage of development; adult stem cells (ASC), which are found in specific tissues and act as a source to repair the damage of their specific tissue; and induced pluripotent stem cells (iPSC), which are adult stem cells that are reprogrammed into another type of adult stem cells and one of the most important cells that can be used for medical purposes. Additionally, there are prenatal stem cells which come from the fetal membrane, umbilical cord, and amniotic fluid extra-embryonic cells, and mesenchymal stem cells (MSC) which are adult stem cells originating from bone marrow, liver, and muscles [2,3][2][3]. For a cell to be considered a stem cell, two criteria must be met. First, stem cells must possess an unlimited capacity for self-renewal in order to produce descendants that are identical to the original cell. Second, they must possess the ability to differentiate into other healthy specialized cells of the body [4]; this specialization can occur depending on the physiological needs of tissue and organs at different times [5].
The cluster regulatory interspaced short palindromic repeats and CRISPR-associated protein 9 (CRISPR-Cas9) system consists of a short and repetitive nucleotide that was first discovered in the genomes of bacteria and archaea that act as adoptive immune systems. It works by removing exogenous genetic elements that assemble with Cas proteins [11][6]. The deactivation of the endogenous genetic elements consists of three steps. First, a few endogenous short nucleotides might integrate with the host’s CRISPR loci as new spacers. Then, a crRNA/Cas complex is created by the transcription of CRISPR RNAs (crRNAs). Finally, under the base complementation pairing rule, the complexes will inactivate the exogenous element [12][7]. Most of the RNase and DNase activities of Cas proteins are predicted with bioinformatics tools. New-generation sequencing has led to the discovery of a large number of Cas proteins; thus, new classification based on sequence information is necessary. The new classification is various and has evolved fast. The new classification has three categories: type 1, type 2 (Cas9 is included in this type and is based on the presence of the HNH domain), and type 3; each type contains a large number of Cas proteins. Despite this classification, some subtypes of Cas proteins are still unclassified. This issue would be improved by further studies in the topic [13,14][8][9]. The type II CRISPR-Cas9 immune system stands out among them because it uses RNase III for cleaving the transcript into mature crRNAs and only needs one Cas9 protein to form a crRNA/Cas9 complex. This technology is simple, fast, cheap, and applicable. These characteristics make it a strong candidate for the development of a completely new genome-editing tool for biological and medical research [15][10]. Gene editing using CRISPR/Cas9 technology has been widely implemented in biological and biomedical research, and stem cell-mediated cell treatment and gene therapy are recognized as essential elements in human medicine because of their capacity for tissue repair and regeneration [16][11]. Some different stem cell types have so far been successfully used in clinical studies and have received scientific approval. Besides viral genome modification, more and more publications are confirming that CRISPR/Cas9 genome editing is a potent technique that can greatly advance biomedicine, such as in virus genome editing and in stem cell research [11,17,18,19][6][12][13][14]. Organoids are interesting new systems that help us improve our knowledge of diseases’ mechanisms, development, evolution, homeostasis, and therapy. They can derive from ESs, ASCs, and PSCs (or even differentiate cells) (Figure 1).

Figure 1. The zygote cell and its initial divisions (embryonic cells), which are considered totipotent stem cells, have the capacity to give rise to fully developed living organisms (body and placenta). Pluripotent stem cells, which can generate all cells that make up a live organism’s body following totipotent stem cells, are the next stage (mesoderm, endoderm, and ectoderm). Organoids are the new era for disease modeling, homeostasis, and development studies. Organoids derived from mesoderm, endoderm, and ectoderm are considered a new field of interest in biomedical research.
2. CRISPR/Cas
2.1. CRISPR/Cas9 and CRISPR/Cas12
CRISPR is a technique that is designed for targeted modification of specific DNA or RNA sequences; nevertheless, it sometimes produces unwanted or unexpected changes in DNA [43][15]. In bacteria and archaebacteria, this mechanism works as an adaptive defense against invading nucleic acids (phages) [44][16]. CRISPR/streptococcus pyogenes CRISPR associated protein 9 (spCas9), the most well-known CRISPR system, is derived from the Streptococcus pyogenes bacterium [45,46,47][17][18][19]. In Streptococcus pyogenes, this system is made up of two primary components. An RNA (containing two distinct RNAs named CRISPR RNA (crRNA) and trans-activator RNA (tracrRNA)) and the Cas9 endonuclease protein, which targets crRNA and tracrRNA [48][20].
Bioinformatics methods were used to create the functional system-related RNA of this bacterium, and instead of two independent components (crRNA and tracrRNA), it has become a single guide RNA (sgRNA). The 20 nucleotides in terminal 5’ of the sgRNA are designed to complement the DNA target location. The presence of a sequence called protospacer adjacent motif (PAM), consisting of 5′-NGG-3′, on the target sites at the end of three of these 20 nucleotides is required for system function [49,50][21][22]. In terms of length and nucleotides, this sequence differs amongst bacteria [51,52,53][23][24][25].
The Cas9 protein identifies this region after the 5’ sgRNA ends of 20 nucleotides bind to the target site. The Cas9 protein then makes a blunt end double-strand break in DNA [51,52,53][23][24][25]. It can take one of two paths after breaking the DNA code—the cell either commits suicide or fixes the damage (Figure 2) [54][26]. NHEJ (non-homologous end joining) and HDR (homology directed repair) are the two main processes for repairing double-stranded breaks in DNA [55][27]. In mammals, the NHEJ mechanism is the most common mode of repair, and most cells use it to mend double-stranded breaks.

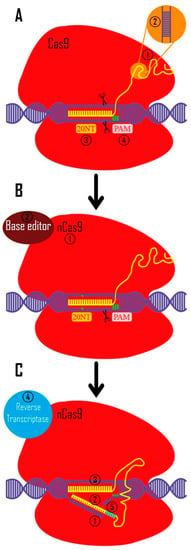
Figure 2. CRISPR toolkit. (A). CRISPR technology was originally used to create double-strand breaks in eukaryotic DNA (with a bacterial origin (Streptococcus pyogenes)). 1. In bacteria, crRNA and tracrRNA guide Cas9 to target the intended region. These RNAs are artificially synthetized as a unique sgRNA to be more applicable in other creatures (yellow) 2. crRNA and tracrRNA are widely used in multiple experimental systems (e.g., mouse embryo microinjections, RNP electroporation into mammalian cell lines, etc.) [91,92][28][29] 3. Twenty nucleotides complementary to the target site are used to identify the target area (these nucleotides are designed in a targeted manner). 4. Before these 20 nucleotides, there are three PAM nucleotides (5′-NGG-3′ in Streptococcus pyogenes Cas9 system) which are necessary for CRISPR/Cas9 function. (B). 1. In order to modify the bases in a targeted way, the Cas9 protein was altered to cut only one strand of DNA by changing one amino acid in Cas9 protein (nickase Cas9 [nCas9]). 2. Additionally, they coupled the different base editor domains to the Cas9 protein. (C). 1. Prime editing, the subsequent iteration of this technique, cuts a DNA strand by creating a cut at the intended location. 2 and 3. The sgRNA is made in such a way that its 3′-end complements the two sides of cut site, and its 5′-end can recognize the target site. 4. The reverse transcriptase enzyme turns 3′ sgRNA into cDNA using this 3′ end as a primer. 5. In the cut region, bases are designed for knock-in to produce highly accurate results.
2.2. Genome-Wide CRISPR/Cas Knock-Out
The genome-scale CRISPR knock-out (GeCKO) method was first developed to examine genes in the human genome [93][30]. With the addition of non-coding areas as new targets for the CRISPR library system, the technology was improved to GeCKO V2 (GeCKO Version 2) [94][31]. This method (genome-wide screening) has been frequently utilized to identify key and fundamental genes involved in diseases [95,96][32][33]. This technique was employed in a study assessing loss of function in iPSC-derived macrophages.
2.3. Dead/Deactivated Cas9 (CRISPR i or a (Inhibition/Activation))
Researchers discovered that D10 (aspartic acid 10) in the RuvC domain and H840 (histidine 840) in the HNH domain are the two amino acids responsible for Cas9 protein double strand DNA cleavage [49][21]. The Cas9 protein loses its capacity to create cuts in the target region when certain amino acids (D10A (aspartic acid 10 alanine) and H840A (histidine 840 alanine)) are changed. Dead/deactivated Cas9 (dCas9) is a modified protein that can only connect to the target site (directed by sgRNA) [100][34]. This protein guides functional domains to the target region chosen by sgRNA by transposing protein functional domains into dCas9. For example, to increase and reduce mRNA expression, dCas9-binding transcription-activator and inhibitor domains are utilized (CRISPR a and I (CRISPR activator and inhibitor)). The sgRNA was created with transcriptional regulatory areas in mind. The functional domain of raising or reducing mRNA transcription is transferred to this area by dCas9. In fact, dCas9 is a carrier of specific functional domains [101,102,103][35][36][37] (e.g., epigenetic regulatory protein domains (acetylase, methylase, deacetylase, demethylase) [104][38], green fluorescence protein (GFP) to identify the DNA locus in the cell [105][39], and base editors to modify the nucleotide without cutting in DNA (Figure 2) [106][40]) and has proved to be a powerful tool of this technology.2.4. RNA Editing
Another kind of this system is CRISPR/Cas13. The only difference is that it breaks RNA rather than DNA [113][41]. Cas13a, b, c, and d are four different types of this system, each with its own set of characteristics [113,114,115][41][42][43]. In CRISPR/Cas13, the area known as PAM in CRISPR/Cas9 and CRISPR/Cas12 is termed as PFS (protospacer flanking site) [115][43]. However, other Cas13 varieties (such as Cas13d) do not require PFS [116][44]. In RNA, this mechanism causes a break [117][45]. Changes to the amino acids in the Cas13 protein have been made, similarly to the CRISPR/Cas9 and CRISPR/Cas12 systems, so that the Cas13 protein binds to the target site only with crRNA guidance. dCas13 gains the ability to effect modifications at the RNA level by binding particular functional domains [118,119,120][46][47][48].2.5. Off-Target
Dedicated sgRNA design is normally carried out using web-based applications [122,123,124][49][50][51]. The most crucial component of a basic design is selecting the optimal sgRNA, created by the software, that has the maximum performance and specificity. Following design, sgRNAs are examined for practical performance in in vitro and in vivo settings, as well as the ability to make cuts (INDEL mutations) using methods such as next-generation sequencing, T7E1, or SURVEYOR kits [125,126][52][53]. Once the best sgRNA has been determined, it will be used for further research. The risk of sgRNA binding to places other than the target site and producing cuts in those regions (off-target) is one of the issues with the CRISPR system [127][54]. Various approaches are employed to prevent these off-target cuts. Double nickase is one of these techniques. The method employs Cas9 systems that have been engineered to cleave a strand of DNA. One dCas9 cuts the sense strand, and the second dCas9 cuts the antisense strand at the same time, resulting in a sticky end double-strand break [128][55]. The off-target cuts are reduced by 50 to 1500 times using this strategy [50][22]. This technique has been utilized to limit the generation of off-target stem cells. R201H has been reported to increase intracellular cGMP production in hPSCs cells utilizing this technology in the guanine nucleotide-binding protein alpha gene stimulating activity polypeptide 1 (GNAS) gene. These cells can be utilized to figure out how the GNAS gene works [129][56]. To test the function of the genes RB1 (retinoblastoma 1) and immune-reactive antigen domain containing 1 (OCIAD1) in iPSCs, this method was employed to generate heterozygous knock-out iPSCs [130,131][57][58].2.6. Knock-In
The knock-in method can be accomplished by a number of ways. All of these techniques are based on using donor DNA to make nucleotide modifications or add a gene or gene cluster to the target site [136][59]. A DNA fragment with a length of 100 to 200 bp can be used to make modifications in several bases. The changed nucleotides are placed in the ssODN middle where the DNA is cut, and the 3′ and 5′ sides of these single stranded oligodeoxynucleotides (ssODNs) are complementary to the two sides of the CRISPR system’s target site. HDR repair is accomplished after DNA cleavage, and the desired nucleotides are altered [137,138][60][61]. Another option is to use prime editing to make these changes. The reverse transcriptase enzyme is coupled to the nickase Cas (nCas) protein in this approach.3. CRISPR Delivery Methods
Delivery methods are classified into two categories: viral vectors and non-viral vectors [158][62]. Safety, low immunogenicity, specific function, minimal toxicity, and high efficiency are all critical properties of a good vector [159,160][63][64]. Adenoviruses, adeno-associated viruses (AAVs), and lentiviruses are the most common viral vectors. These viruses are being used in a number of clinical investigations [159][63]. Lentiviral vectors can transport larger amounts of DNA, and the third generation of these viruses is undergoing clinical trials [161,162][65][66]. Additionally, these viruses have the ability to penetrate cells efficiently, produce a large number of in vitro viruses, and they have a high cell transduction rate [163,164,165][67][68][69]. However, one of the most essential characteristics of viruses is their capacity to integrate into host DNA. AAVs were found, in a 10-year long-term study, to have the ability to integrate into DNA and induce cancer in dogs with hemophilia who have been treated with the virus [166][70]. The virus can enter the genome through the CRISPR system’s cut area and remain there for a long time [167,168][71][72]. To overcome this problem, virus-like particles that do not have the ability to integrate their genome into the host’s genome are used. This delivery method is used for both in vitro and in vivo editing. These viruses are non-proliferative and have good delivery efficiency [169][73]. The PiggyBac transposon system is the other technology for gene(s) or cluster delivery.References
- Zakrzewski, W.; Dobrzynski, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68.
- Mummery, C.; van de Stolpe, A.; Roelen, B.A.J.; Clevers, H. (Eds.) Chapter 5—Origins and Types of Stem Cells: What’s in a Name? In Stem Cells, 2nd ed.; Academic Press: Boston, MA, USA, 2014; pp. 101–129.
- Razazian, M.; Khosravi, M.; Bahiraii, S.; Uzan, G.; Shamdani, S.; Naserian, S. Differences and similarities between mesenchymal stem cell and endothelial progenitor cell immunoregulatory properties against T cells. World J. Stem Cells 2021, 13, 971–984.
- Biehl, J.K.; Russell, B. Introduction to Stem Cell Therapy. J. Cardiovasc. Nurs. 2009, 24, 98–103.
- Singh, V.K.; Saini, A.; Kalsan, M.; Kumar, N.; Chandra, R. Describing the Stem Cell Potency: The Various Methods of Functional Assessment and In silico Diagnostics. Front. Cell Dev. Biol. 2016, 4, 134.
- Valenti, M.T.; Serena, M.; Carbonare, L.D.; Zipeto, D. CRISPR/Cas system: An emerging technology in stem cell research. World J. Stem Cells 2019, 11, 937–956.
- Kunin, V.; Sorek, R.; Hugenholtz, P. Evolutionary conservation of sequence and secondary structures in CRISPR repeats. Genome Biol. 2007, 8, R61.
- Makarova, K.; Wolf, Y.; Koonin, E. Classification and nomenclature of CRISPR-cas systems: Where from here? CRISPR J. 2018, 1, 325–336.
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and classification of the CRISPR–Cas systems. Nat. Rev. Genet. 2011, 9, 467–477.
- Zhang, Z.; Zhang, Y.; Gao, F.; Han, S.; Cheah, K.S.; Tse, H.-F.; Lian, Q. CRISPR/Cas9 Genome-Editing System in Human Stem Cells: Current Status and Future Prospects. Mol. Ther.-Nucleic Acids 2017, 9, 230–241.
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264.
- Jamehdor, S.; Pajouhanfar, S.; Saba, S.; Uzan, G.; Teimoori, A.; Naserian, S. Principles and Applications of CRISPR Toolkit in Virus Manipulation, Diagnosis, and Virus-Host Interactions. Cells 2022, 11, 999.
- Jamehdor, S.; Zaboli, K.A.; Naserian, S.; Thekkiniath, J.; Omidy, H.A.; Teimoori, A.; Johari, B.; Taromchi, A.H.; Sasano, Y.; Kaboli, S. An overview of applications of CRISPR-Cas technologies in biomedical engineering. Folia Histochem. Cytobiol. 2020, 58, 163–173.
- Jamehdor, S.; Naserian, S.; Teimoori, A. Enhanced High Mutation Rate and Natural Selection to Produce Attenuated Viral Vaccine with CRISPR Toolkit in RNA Viruses especially SARS-CoV-2. Infect Genet Evol. 2022, 97, 105188.
- Yan, F.; Wang, W.; Zhang, J. CRISPR-Cas12 and Cas13: The lesser known siblings of CRISPR-Cas9. Cell Biol. Toxicol. 2019, 35, 489–492.
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190.
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature 2017, 550, 407–410.
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495.
- Le Rhun, A.; Escalera-Maurer, A.; Bratovič, M.; Charpentier, E. CRISPR-Cas in Streptococcus pyogenes. RNA Biol. 2019, 16, 380–389.
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308.
- Hou, Z.; Zhang, Y.; Propson, N.E.; Howden, S.E.; Chu, L.-F.; Sontheimer, E.J.; Thomson, J.A. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc. Natl. Acad. Sci. USA 2013, 110, 15644–15649.
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Topkar, V.V.; Nguyen, N.T.; Zheng, Z.; Gonzales, A.P.W.; Li, Z.; Peterson, R.T.; Yeh, J.-R.J.; et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 2015, 523, 481–485.
- Tóth, E.; Weinhardt, N.; Bencsura, P.; Huszár, K.; Kulcsár, P.I.; Tálas, A.; Fodor, E.; Welker, E. Cpf1 nucleases demonstrate robust activity to induce DNA modification by exploiting homology directed repair pathways in mammalian cells. Biol. Direct 2016, 11, 46.
- Featherstone, C.; Jackson, S.P. DNA double-strand break repair. Curr. Biol. 1999, 9, R759–R761.
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714.
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53.
- Ma, X.; Chen, C.; Veevers, J.; Zhou, X.; Ross, R.S.; Feng, W.; Chen, J. CRISPR/Cas9-mediated gene manipulation to create single-amino-acid-substituted and floxed mice with a cloning-free method. Sci. Rep. 2017, 7, 42244.
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science 2014, 343, 84–87.
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784.
- Baggen, J.; Persoons, L.; Vanstreels, E.; Jansen, S.; Van Looveren, D.; Boeckx, B.; Geudens, V.; De Man, J.; Jochmans, D.; Wauters, J.; et al. Genome-wide CRISPR screening identifies TMEM106B as a proviral host factor for SARS-CoV-2. Nat. Genet. 2021, 53, 435–444.
- Zhu, Y.; Feng, F.; Hu, G.; Wang, Y.; Yu, Y.; Zhu, Y.; Xu, W.; Cai, X.; Sun, Z.; Han, W.; et al. A genome-wide CRISPR screen identifies host factors that regulate SARS-CoV-2 entry. Nat. Commun. 2021, 12, 961.
- Brezgin, S.; Kostyusheva, A.; Kostyushev, D.; Chulanov, V. Dead Cas Systems: Types, Principles, and Applications. Int. J. Mol. Sci. 2019, 20, 6041.
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196.
- MacLeod, R.S.; Cawley, K.M.; Gubrij, I.; Nookaew, I.; Onal, M.; O’Brien, C.A. Effective CRISPR interference of an endogenous gene via a single transgene in mice. Sci. Rep. 2019, 9, 17312.
- Schoger, E.; Argyriou, L.; Zimmermann, W.-H.; Cyganek, L.; Zelarayán, L.C. Generation of homozygous CRISPRa human induced pluripotent stem cell (hiPSC) lines for sustained endogenous gene activation. Stem Cell Res. 2020, 48, 101944.
- Pulecio, J.; Verma, N.; Mejía-Ramírez, E.; Huangfu, D.; Raya, A. CRISPR/Cas9-based engineering of the epigenome. Cell Stem Cell 2017, 21, 431–447.
- Chen, B.; Huang, B. Imaging Genomic Elements in Living Cells Using CRISPR/Cas9. Methods Enzymol. 2014, 546, 337–354.
- Eid, A.; AlShareef, S.; Mahfouz, M.M. CRISPR base editors: Genome editing without double-stranded breaks. Biochem. J. 2018, 475, 1955–1964.
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573.
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027.
- Konermann, S.; Lotfy, P.; Brideau, N.J.; Oki, J.; Shokhirev, M.N.; Hsu, P.D. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 2018, 173, 665–676.e14.
- Yan, W.X.; Chong, S.; Zhang, H.; Makarova, K.S.; Koonin, E.V.; Cheng, D.R.; Scott, D.A. Cas13d is a compact RNA-targeting type VI CRISPR effector positively modulated by a WYL-domain-containing accessory protein. Molecular cell 2018, 70, 327–339.e325.
- Granados-Riveron, J.T.; Aquino-Jarquin, G. CRISPR–Cas13 Precision Transcriptome Engineering in Cancer. Cancer Res. 2018, 78, 4107–4113.
- Li, J.; Chen, Z.; Chen, F.; Xie, G.; Ling, Y.; Peng, Y.; Lin, Y.; Luo, N.; Chiang, C.-M.; Wang, H. Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein. Nucleic Acids Res. 2020, 48, 5684–5694.
- Shimizu, Y.; Bandaru, S.; Hara, M.; Young, S.; Sano, T.; Usami, K.; Kurano, Y.; Lee, S.; Kumagai-Takei, N.; Sano, S.; et al. An RNA-immunoprecipitation via CRISPR/dCas13 reveals an interaction between the SARS-CoV-2 5’UTR RNA and the process of human lipid metabolism. Res. Sq. 2021; Preprint.
- Crunkhorn, S. Expanding the RNA-editing toolbox. Nat. Rev. Drug Discov. 2019, 18, 667.
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80.
- Keough, K.C.; Lyalina, S.; Olvera, M.P.; Whalen, S.; Conklin, B.R.; Pollard, K.S. AlleleAnalyzer: A tool for personalized and allele-specific sgRNA design. Genome Biol. 2019, 20, 167.
- Liu, H.; Wei, Z.; Dominguez, A.; Li, Y.; Wang, X.; Qi, L.S. CRISPR-ERA: A comprehensive design tool for CRISPR-mediated gene editing, repression and activation: Figure 1. Bioinformatics 2015, 31, 3676–3678.
- Dong, Y.; Li, H.; Zhao, L.; Koopman, P.; Zhang, F.; Huang, J.X. Genome-Wide Off-Target Analysis in CRISPR-Cas9 Modified Mice and Their Offspring. G3 Genes Genomes Genet. 2019, 9, 3645–3651.
- Zischewski, J.; Fischer, R.; Bortesi, L. Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 2017, 35, 95–104.
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e264.
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389.
- Watanabe, K.; Nakamura, T.; Onodera, S.; Saito, A.; Shibahara, T.; Azuma, T. A novel GNAS-mutated human induced pluripotent stem cell model for understanding GNAS-mutated tumors. Tumor Biol. 2020, 42, 1010428320962588.
- Shetty, D.K.; Kalamkar, K.P.; Inamdar, M.S. OCIAD1 controls electron transport chain complex I activity to regulate energy metabolism in human pluripotent stem cells. Stem Cell Rep. 2018, 11, 128–141.
- Tu, J.; Huo, Z.; Liu, M.; Wang, D.; Xu, A.; Zhou, R.; Zhu, D.; Gingold, J.; Shen, J.; Zhao, R.; et al. Generation of human embryonic stem cell line with heterozygous RB1 deletion by CRIPSR/Cas9 nickase. Stem Cell Res. 2018, 28, 29–32.
- Banan, M. Recent advances in CRISPR/Cas9-mediated knock-ins in mammalian cells. J. Biotechnol. 2019, 308, 1–9.
- Bialk, P.; Rivera-Torres, N.; Strouse, B.; Kmiec, E.B. Regulation of Gene Editing Activity Directed by Single-Stranded Oligonucleotides and CRISPR/Cas9 Systems. PLoS ONE 2015, 10, e0129308.
- Okamoto, S.; Amaishi, Y.; Maki, I.; Enoki, T.; Mineno, J. Highly efficient genome editing for single-base substitutions using optimized ssODNs with Cas9-RNPs. Sci. Rep. 2019, 9, 4811.
- Sung, Y.K.; Kim, S.W. Recent advances in the development of gene delivery systems. Biomater. Res. 2019, 23, 8.
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021, 6, 53.
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy- an overview. J. Clin. Diagn Res. 2015, 9, GE01-6.
- Loza, L.I.M.; Yuen, E.C.; McCray, J.P.B. Lentiviral Vectors for the Treatment and Prevention of Cystic Fibrosis Lung Disease. Genes 2019, 10, 218.
- Negre, O.; Eggimann, A.V.; Beuzard, Y.; Ribeil, J.A.; Bourget, P.; Borwornpinyo, S.; Hongeng, S.; Hacein-Bey, S.; Cavazzana, M.; Leboulch, P.; et al. Gene Therapy of the beta-Hemoglobinopathies by Lentiviral Transfer of the beta(A(T87Q))-Globin Gene. Hum. Gene Ther. 2016, 27, 148–165.
- Ghaleh, H.E.G.; Bolandian, M.; Dorostkar, R.; Jafari, A.; Pour, M.F. Concise review on optimized methods in production and transduction of lentiviral vectors in order to facilitate immunotherapy and gene therapy. Biomed. Pharmacother. 2020, 128, 110276.
- Ricobaraza, A.; Gonzalez-Aparicio, M.; Mora-Jimenez, L.; Lumbreras, S.; Hernandez-Alcoceba, R. High-Capacity Adenoviral Vectors: Expanding the Scope of Gene Therapy. Int. J. Mol. Sci. 2020, 21, 3643.
- Westhaus, A.; Cabanes-Creus, M.; Rybicki, A.; Baltazar, G.; Navarro, R.G.; Zhu, E.; Drouyer, M.; Knight, M.; Albu, R.F.; Ng, B.H.; et al. High-Throughput In Vitro, Ex Vivo, and In Vivo Screen of Adeno-Associated Virus Vectors Based on Physical and Functional Transduction. Hum. Gene Ther. 2020, 31, 575–589.
- Kaiser, J. Virus used in gene therapies may pose cancer risk, dog study hints. Science 2020, 10.
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439.
- Nelson, C.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Rivera, R.M.C.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432.
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 2022, 185, 250–265.e16.
More