Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Siheun Lee and Version 2 by Rita Xu.
Cell-to-cell variation exists within a population of the same cell type due to stochastic gene and protein expression and environmental factors. Studying such cellular heterogeneity is the key to understanding the underlying mechanisms of fundamental biology and complex diseases, highly demanding developments in advanced technologies for molecular profiling at the single-cell level.
- mass spectrometry
- single-cell analysis
- proteomics
1. Introduction
The heterogeneity of cells in populations caused by cell-to-cell variation makes it necessary to analyze single cells and this will allow one to discover hidden mechanisms not seen in bulk samples (Figure 1) [1][2][1,2]. Recent studies utilizing both antibody-based methods and mass spectrometry-based methods successfully demonstrated the importance of single-cell analysis [3][4][3,4]. These single-cell analysis methods have been making rapid progress in terms of higher sensitivity as well as increased identification numbers and specificity. Single-cell analysis is essential for a deeper understanding of cancer, immunology, and other fields requiring precise information on cellular mechanisms. Diverse molecules in cells, such as RNA, proteins, and metabolites, can be analyzed at the single-cell level.
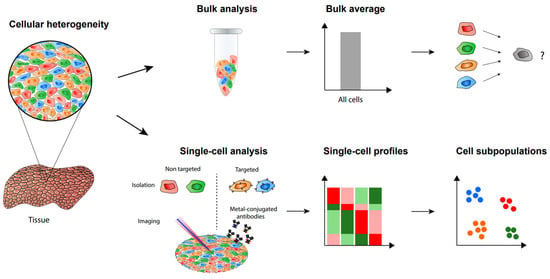
Figure 1. Single-cell analysis and imaging reveal cellular heterogeneity not seen by bulk analysis methods.
Among common approaches for single cell protein analysis are antibody-based methods. They are characterized using specific antibodies that bind to target proteins, which can then be identified through several techniques. Immunocytochemistry (ICC) is one such approach in which cultured cells or individual cells that have been isolated are tagged using an antibody of interest [5][6][5,6]. The antibody is linked to a reporter, usually a fluorophore or enzyme, which can then be detected in a microscope after fluorescence or color from an enzymatic reaction occurs. Another immunofluorescence method is immunohistochemistry (IHC), which differs from ICC in the fact that the cell staining is applied to intact tissue sections [7]. With ICC, most of the extracellular matrix and interstitial components are removed, leaving only isolated cells to be analyzed. For the conventional immunofluorescence analysis of single cells, ICC is generally used as it includes a cell isolation procedure. However, a comparison of the results between ICC and IHC may provide some insight into the differences between single cells and bulk tissue samples regarding the distribution of specific antigens. Another widely used method is fluorescence activated cell sorting (FACS). This method utilizes laser-induced fluorophores to count cells of interest based on antibody-antigen interaction. It has been reviewed elsewhere [8][9][8,9] and will not be discussed here. One critical limitation of antibody-based methods is that there must be antigen-specific binding between the target protein and antibody, which requires the rigorous testing of specificity [6].
Next-generation sequencing (NGS) has also proven to be a powerful tool in the analysis of single cells [10][11][10,11]. Whole genomes can be sequenced within a day and the increased sensitivity can lead to the detection of genetic alterations, such as somatic variants. In addition, RNA sequencing (RNA-Seq) can be used for the discovery of novel RNA variants and splice sites, as well as the quantification of mRNAs for gene expression analysis [12]. Single-cell RNA sequencing (scRNA-seq) allows for new biological discoveries, which otherwise would be unobtainable using traditional methods that analyze pooled bulk RNAs from tissues. These include the identification of rare cell types [13][14][13,14], gene regulatory networks inference [15][16][17][15,16,17], and cell type hierarchy reconstruction [18][19][18,19].
Mass spectrometry (MS) has been an essential tool for analyzing single cells [20][21][20,21]. Although it is the most powerful method for protein analysis, there have been challenges in its application to single cells. However, advances in simple, multiplexed, automated, and scaled-down sample preparation have opened doors for rapid analysis with high sensitivity. Along with single-cell proteomics, MS has also been used to identify and quantify metabolites and lipids at the single-cell level.
2. Label-Free Single-Cell Proteomic Analysis
Liquid chromatography tandem mass spectrometry analysis (LC-MS/MS) is one of the most effective methods in proteome profiling at the single-cell level. The rapid evolution of the LC-MS system over recent years has enabled label-free single-cell proteomic (SCP) analysis capable of identifying and quantifying more than 1000 proteins [22][23][24][22,23,24]. The current platforms for label-free SCP analysis were mainly developed with either Orbitrap or trapped ion mobility spectrometry time of flight (timsTOF) mass spectrometers. Cong et al. recently introduced an ultra-sensitive workflow that combines nanoPOTS (nanodroplet processing in one pot for trace samples), ultra-low flow liquid chromatography, and high field asymmetric ion mobility spectrometry (FAIMS) with an Orbitrap Eclipse Tribrid mass spectrometer [23]. The workflow enhanced the depth of single-cell proteome with 1056 identified proteins on average. Importantly, the employment of FAIMS has tripled the number of identified proteins compared to their previous research, indicating the critical contribution of ion mobility technology to the gas-phase separation [25]. The benefit of FAIMS was further elevated in a new method named transferring identification based on FAIMS filtering (TIFF) [24]. Besides the mass-to-charge ratio value (m/z) and the retention time, TIFF utilized the FAIMS compensation voltage (CV) as a third-dimensional characteristic of precursor ions for the peptide identification of single cells based on the match between run (MBR) algorithm. The efficiency of TIFF was demonstrated with the average of proteome coverage increased to over 1200 proteins. Together with FAIMS-Orbitrap mass spectrometer system, timsTOF mass spectrometer, with its remarkable sensitivity, has been used in several label-free SCP analyses [22][26][22,26]. Newly introduced timsTOF for single-cell proteomic analysis showed ten-fold increased sensitivity with the optimal setting, enabling the identification of 843 proteins on average in a single Hela cell [22]. Data dependent acquisition (DDA) is the most common scan mode in LC-MS/MS analysis [27][28][29][30][31][27,28,29,30,31]. DDA methods have been initially used for single cell proteomics analysis [23][25][32][23,25,32]. However, the DDA strategy only selects a small percent of precursors ion for tandem mass analysis, leading to low data completeness in numerous samples such as single cells. Data-independent acquisition (DIA) method recently gained attraction in label-free proteomics analysis on account of minimal missing values across replicates [33][34][33,34]. A new microfluid chip named SciProChip was developed for SCP analysis in DIA mode by Gebreyesus and his colleagues [35]. Applying SciProChip to DIA analysis resulted in the identification of approximately 1500 proteins on average from single cells with less than 16% missing values. In the timsTOF SCP, diaPASEF (parallel accumulation-serial fragmentation combined with data-independent acquisition) scan mode was established to maximize the number of precursor ions for tandem mass spectrometry [32]. This method has increased the number of quantifiable proteins (up to 2083) per single Hela cell with high completeness [22]. Although the proteome coverage in label-free SCP analysis remains low, several important cellular biological processes were observed. Proteome profiling from single PC9 cells showed several proteins involved in NSCLC pathways, such as EGFR, TP53, NRAS and MAPK [35]. In another research, proteins related to cell cycles were readily quantifiable in SCP data with several differentially expressed proteins upon drug treatment [32]. These results demonstrated the capability of using label-free SCP analysis in biological and clinical applications in the future.3. TMT-Assisted Single-Cell Proteomics
Tandem mass tags (TMT) are isobaric chemicals used for the accurate and multiplexed quantification of peptides and proteins using tandem MS analysis (Figure 2) [29][36][37][38][39][29,36,37,38,39]. All multiple reagents of a typical TMT set have the same nominal mass and an identical chemical structure composed of a mass reporter, a mass normalizer, and an amine-reactive group (Figure 2a). Each mass reporter of the reagents contains stable isotopes distinctly configured in the chemical structure, thus having different masses from one another. For TMT-based multiplexed proteomics, digested peptides from different samples are labeled with TMT reagents (10, 16, or 18-plex [40]) and analyzed simultaneously by a single run of LC-MS/MS. The mass reporters are cleaved from the TMT-labeled peptides after fragmentation and distinguished based on their distinct masses specific to the samples. Their ion intensities measured in MS2 spectrum and corresponding peptide sequencing enables relative quantification of the peptides. Multiplexed analysis using TMT alleviates the variability of separate measurements, and the enhanced intensity of precursor ions, which are accumulated from identical TMT-labeled peptides of all samples, improves the quantification of proteins [41]. The labeling method with TMT detected changes in the number of low-abundance proteins available for hypothesis testing, showing higher precision and fewer missing values compared to a label-free quantitation method [42]. It should be noted that the accurate interpretation of TMT-based quantitative proteomic data requires minimizing false positives, batch effects, and missing values [43][44][43,44].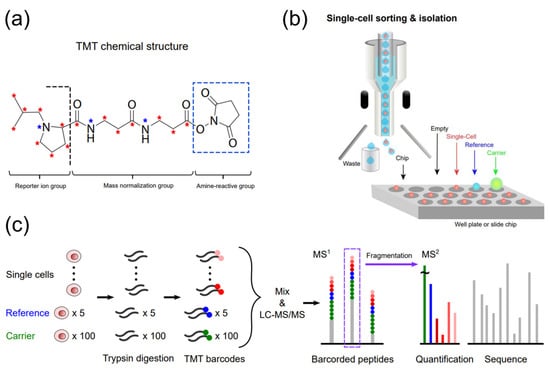
Figure 2. TMT-assisted single-cell proteomics. (a) The chemical structure of TMT (e.g., 18-plex), (b) Single-cell sorting and isolation onto a well plate or slide chip by FACS or a robotic system. (c) A typical workflow of single-cell proteomic analysis using TMT.
4. CyTOF for Single-Cell Proteomics
Cytometry by time-of-flight (CyTOF) (Figure 3), or mass cytometry, is a variation of flow cytometry. Flow cytometry is a widely used immune profiling method in which a sample of cells is labeled with fluorescent markers and examined using a laser beam [69][70][71][69,70,71]. Each cell from the sample flows in a row through the laser excitation region, where multiple fluorescent markers of the single cells are measured simultaneously, and the fluorescence intensities represent a proxy of the expression level of the targeted antigens. Despite a high-throughput method used to analyze or quantify multiple cellular features at the single-cell level, there are limitations with flow cytometry. Fluorescence measurements often create spectral overlap and require compensation, limiting the number of parameters that can be measured together [72].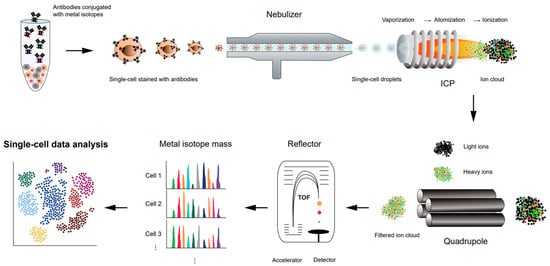
Figure 3. Schematic representation of a workflow of high-throughput single-cell proteomics analysis using CyTOF.
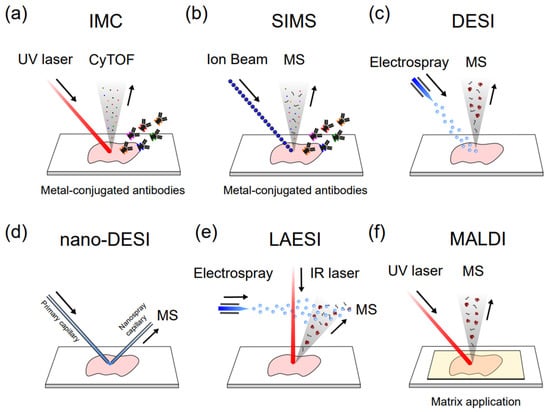
Figure 4. Different single-cell MSI methods. (a) Imaging mass cytometry (IMC); (b) Secondary ion mass spectrometry (SIMS); (c) Desorption electrospray ionization (DESI); (d) nanospray-DESI (nano-DESI); (e) Laser ablation electrospray ionization (LAESI); (f) Matrix-assisted laser desorption/ionization.