Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Mohammad Al-Mahdi Al-Karagholi and Version 3 by Rita Xu.
Involvement of glutamate and its peripheral N-Methyl-D-Aspartate Receptor (NMDAR) in migraine pathophysiology. N-Methyl-D-Aspartate Receptor (NMDAR) are expressed at several levels of the trigeminovascular system, which is the anatomical and physiological substrate of migraine pain.
Involvement of glutamate and its peripheral N-Methyl-D-Aspartate Receptor (NMDAR) in migraine pathophysiology.
- glutamate
- headache
- pain
- aura
1. Introduction
Migraine is a primary headache disorder, affecting more than 15% of the global adult population in their most productive years of life with a health and economic burden of billions of dollars globally [1]. The clinical manifestation of migraine is recurrent attacks with a severe, and usually unilateral and throbbing headache, lasting 4–72 h and associated with nausea and/or light and sound sensitivity [2]. In one-third of individuals with migraine, the headache phase is preceded by transient focal neurological disturbances, the so-called migraine aura-phase [3], whose underlying mechanism is considered to be cortical spreading depression (CSD) [4]. CSD is a self-sustaining, slowly propagating wave of transient neuronal and glial depolarization [3] that silences brain and electrical activity for several minutes [5][6][7][5,6,7]. In the past three decades, advances made in migraine research led to the development of several acute and preventive medications. Yet, a significant proportion of patients reports an inadequate response and a lack of tolerability [8].
Glutamate is the principal excitatory neurotransmitter in the central nervous system [9], and its role in migraine pathophysiology has been under the spotlight for more than three decades [10][11][12][13][10,11,12,13]. Glutamate receptors are pharmacologically classified as ionotropic and metabotropic receptors. N-Methyl-D-Aspartate Receptor (NMDAR) is an ionotropic glutamate receptor [9] that modulates excitatory neurotransmission by conducting sodium (Na+) and calcium (Ca2+) ions into the cell and potassium (K+) ions outside the cell. Key structures related to migraine pain, including the trigeminal ganglion (TG), trigeminal nucleus caudalis (TNC), and thalamus contain high densities of NMDAR-positive neurons, pinpointing a potential connection between the NMDAR and migraine pathophysiology [9][12][14][15][16][9,12,14,15,16].
2. N-Methyl-D-Aspartate Receptor (NMDAR)
Glutamate receptors are pharmacologically classified as ionotropic (ligand-gated ion channels) and metabotropic (GPCRs). NMDAR along with alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor (AMPAR) and Kainate receptor (KAR) are categorized as ionotropic glutamate receptors [9]. NMDAR modulates excitatory neurotransmission by conducting Na+ and Ca2+ ions into the cell and K+ ions outside the cell (Figure 1). The NMDAR is a heterotetrameric complex assembled from three different NMDAR subunits (NR1, NR2, and NR3) [17][35] (Figure 2). The NR1 subunit is essential for a functional NMDAR complex [17][18][35,36] and is represented by eight splice variants, which influence the channel properties. Four NR2 subunits (NR2A, NR2B, NR2C and NR2D) and two NR3 subunits (NR3A and NR3B) have been mapped. The NR1-NR2A heterodimer suggests a mechanism for ligand-induced ion channel opening and is the functional unit in tetrameric NMDARs [19][37]. All NMDAR subunits share a common membrane structure characterized by a large N-terminus domain (NTD), an agonist/ligand binding domain (ABD/LBD), a transmembrane region (TMD), and a cytoplasmic region (CTD) (Figure 2) [17][35]. NMDAR activation requires the simultaneous binding of glycine and glutamate, together with the removal of the endogenous channel-pore blocker, magnesium ion (Mg2+), in a voltage-dependent manner [20][21][38,39]. The particular importance of the NMDAR is due to the high permeability to Ca2+ ions that gives NMDARs a significant role in both synaptic plasticity under physiological conditions and neuronal death under excitotoxic pathological conditions [17][35]. A typical NMDAR complex consists of two glycine-binding NR1 subunits and two glutamate-binding NR2 subunits [18][36]. Incorporating either NR3A or NR3B subunits showed decreased channel activity via reduced single-channel conductance, reduced Ca2+ permeability and increased Mg2+ blockade [17][22][23][24][35,40,41,42]. Notably, the NR3 subunit can bind to glycine rather than glutamate, which is similar to the NR1 subtype. Thus, a novel type of NMDAR complex composed of NR1 and NR3 subunits would only require glycine and not glutamate for activation [25][43]. The entry of Ca2+ into dendritic spines through NMDARs is essential for long-term potentiation (LTP) and long-term depression (LTD) [26][44]. LTP is induced by a high-frequency synaptic activity that causes postsynaptic membrane depolarization, a decrease in voltage-dependent Mg2+ blockage of the NMDAR pore, and a massive entry of Ca2+ ions into dendritic spines leading to calmodulin (CaM) and CaM-dependent kinase II activation (Figure 1).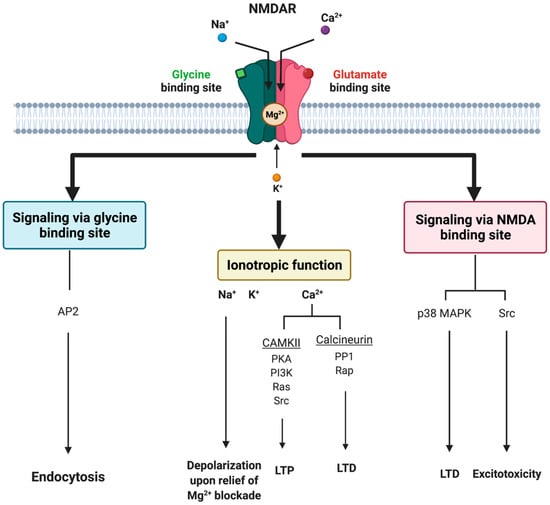
Figure 1. Tripartite signaling of N-Methyl-D-Aspartate Receptor (NMDAR). This model illustrates three parallel signaling pathways of NMDAR transduction. Binding of glutamate and glycine results in ionotropic function and membrane depolarization through efflux of K+ ions and influx of Na+ and Ca2+ ions mediating downstream Ca2+-dependent pathways. NMDAR signaling can also happen non-ionotropically when glycine or glutamate bind independently and induce conformational changes and downstream protein interactions. Glycine stimulation enhances NMDAR association with the AP2 and subsequent activation of a downstream endocytic pathway. Lastly, glutamate binding can result in LTD through p38 MAPK and excitotoxicity though an activation of Src. AP2 = endocytic adaptor protein 2, CaMKII = calcium/calmodulin kinase II, LTD = long-term depression, LTP = long-term potentiation, p38 MAPK = p38 mitogen-activated protein kinase, PP1 = protein phosphatase 1, Rap = ras-related protein, Src = thyrosin kinase family.
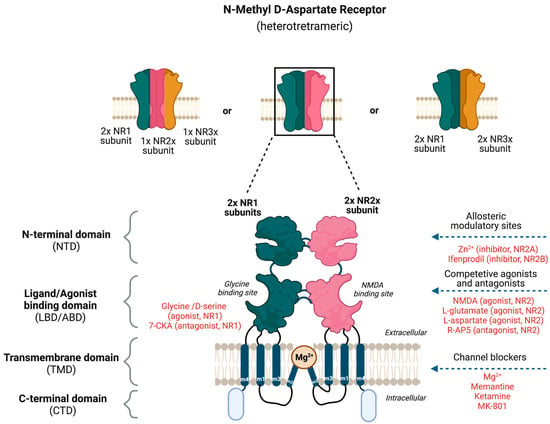
Figure 2. Potential binding sites for ligands that modulate the activity of N-Methyl-D-Aspartate Receptor (NMDAR). The NMDAR is assembled as tetramers; two NR1 and two NR2 subunits forming a dimer–dimer structure or two NR1 subunits associating with one NR2 and NR3 subunit. The channel construction with different domains gives rise to ligand binding or allosteric modulatory sites that either work as an NMDAR agonist or antagonist.