It is now well known that the bone marrow (BM) cell niche contributes to leukemogenesis, but emerging data support the role of the complex crosstalk between AML cells and the BM microenvironment to induce a permissive immune setting that protects leukemic stem cells (LSCs) from therapy-induced death, thus favoring disease persistence and eventual relapse. The identification of potential immune targets on acute myeloid leukemia (AML) cells and the modulation of the BM environment could lead to enhanced anti-leukemic effects of drugs, immune system reactivation, and the restoration of AML surveillance. Potential targets and effectors of this immune-based therapy could be monoclonal antibodies directed against LSC antigens such as CD33, CD123, and CLL-1 (either as direct targets or via several bispecific T-cell engagers), immune checkpoint inhibitors acting on different co-inhibitory axes (alone or in combination with conventional AML drugs), and novel cellular therapies such as chimeric antigen receptor (CAR) T-cells designed against AML-specific antigens.
- acute myeloid leukemia
- drug resistance
- immune escape
- immune therapy
1. Introduction
2. Leukemic Cells in Their Bone Marrow Microenvironment: How They Induce Drug Resistance and Escape Immune Response
A hierarchical organization of bone marrow leukemic cells resembling the normal hematopoietic system, with a small subset of leukemic stem cells (LSCs) on the top of the pyramid, continually replenishing the more mature bulky population, was first described 25 years ago by Bonnet and coll. [5][8]. As for the normal counterpart, leukemic stem cells are defined by their self-renewal capacity and their ability to initiate leukemia after serial transplantation in SCID mice [5][6][8,9]. Interestingly, LSCs seem to originate from the acquisition of driver mutations, not by normal immature hematopoietic stem cells (HSCs) but by committed progenitors. At least two distinct LSC populations have been identified in human AMLs: a more mature population, emerging from granulocyte-macrophage progenitors (GMPs), and a more immature population, from lymphoid-primed multipotent progenitors (LMMPs) [7][10]. The recent identification of the clonal hematopoiesis of indeterminate potential (CHIP) [8][11] and the recognized ability of more mature LSCs to revert to an immature state [9][12] have further complicated the scenario. However, the persistence of pre-leukemic hematopoietic clonal alteration in responding patients confirms the negative role of LSCs [10][13]. Although it is well known that LSCs are enriched within the CD34+CD38- fraction, a clear definition of the LSC phenotype does not exist. Compared to normal HSCs, LSCs may have higher expression of CD25, CD32, CD44, CD96, CD123, CD200, GPR56, N-cadherin, Tie2, TIM-3, CLL-1, c-MPL, and HDM2 [11][12][13][14][15][16][17][18][19][20][21][14,15,16,17,18,19,20,21,22,23,24]. Unfortunately, the high inter- and intra-individual heterogeneity complicates their enumeration and sequential monitoring. Despite this, the negative prognostic role of LSC frequency at diagnosis or after therapy has been demonstrated by many studies, and different LSC prognostic scores have been obtained by gene expression profiles and whole exome plantation [22][23][24][25,26,27]. LSCs escape the effect of anti-leukemic drugs by nesting in the BM microenvironment in a quiescent state, and leukemic progeny gradually occupy the BM niche, converting it into a “leukemic niche”, able to support leukemic cells’ survival and proliferation while decreasing its capacity to maintain normal hematopoiesis [25][26][27][28,29,30]. It is interesting to underline that BM failure is not due to a reduction in HSCs but to its inability to produce an adequate number of progenitors as a consequence of the release by mesenchymal cells (MSCs) of hypoxia-associated molecules that increase stemness and prevent differentiation [28][31], mimicking the differentiation arrest that is the hallmark of the leukemia phenotype. The modifications of the BM niche by leukemic cells involve all the components of the microenvironment. The endosteal niche is reshaped by the loss of balance between bone formation and resorption as the consequence of the activation of the RANK/RANKL pathway, which promotes osteoclastogenesis and favors osteoclast survival [29][32]. Moreover, the secretion by LSCs of a bone morphogenic protein (BMP), of a negative regulator of osteogenesis (DKK), and of a chemokine that decreases osteocalcin and switches MSC differentiation from adipogenic to osteoblastic with the consequent accumulation of progenitor and immature osteoblasts and defects in bone mineralization (CCL3, also known as MIP1α) [30][31][33,34], contribute to creating a milieu of facilitating leukemia cell growth and AML progression. Furthermore, in vitro data suggest that MSCs also support leukemia cell survival and promote resistance to chemotherapy, inducing a low cycling rate and anti-apoptotic signals [32][33][35,36]. In the AML vascular niche, increased IL-1β and TNF-α levels synergize with increased adhesive receptors E-selectin/CD44 and VCAM-1/VLA-4, thus ensuring the anchorage of LSCs to endothelial cells [34][35][37,38]. VEGF/VEGFR and Notch/Delta-like ligand 4 promote neo-angiogenesis, essential for AML progression and extramedullary homing [36][37][39,40]. Although less known compared to the other, the reticular niche is considered a transitional niche, able to maintain HSCs in a proliferative state, such as the vascular, but in an undifferentiated state, such as the endosteal one. CXCL12-abundant reticular (CAR) cells and Nestin-expressing cells are the most represented cells in the niche, localized in proximity to sinusoidal endothelial cells [38][39][40][41,42,43]. Most HSCs are in contact with CAR cells, but interaction with these cells is established also by B-lymphocytes, plasma cells, plasmacytoid dendritic cells, and NK cells, suggesting that reticular cells might also act as an immune cells niche [41][42][43][44,45,46]. CAR cells secrete CXCL12 and SCF, crucial for HSCs trafficking and homing. In AML, the CXCL12/CXCR4 axis regulates the infiltration of leukemic cells in the protective bone marrow niche [44][45][47,48]. Nestin-expressing cells are associated with adrenergic nerves and regulate HSC maintenance [40][46][43,49]. AML bone marrow is enriched with Nestin-expressing cells. Besides their role in developing AML, Nestin-expressing cells are implicated in the induction of resistance to chemotherapy by enhanced glutathione (GSH)-peroxidase (Gpx) activity [47][50].Considered in the past only as passive bystanders, BM adipocytes actually play an important role in regulating normal HSCs and are implicated in hematopoietic recovery after irradiation or 5FU treatment by secreting SCF [48][51]. In AML, they promote leukemic cell survival during therapy and drug resistance by enhancing free fatty acid production [49][50][52,53] and by sequestering, inactivating, and metabolizing chemotherapeutic drugs [51][54]. Among cells from myeloid resident in the bone marrow, myeloid-derived suppressor cells (MDSCs) and leukemia-associated macrophages (LAMs) are also implicated in generating an immunosuppressive microenvironment in AML. MDSCs are a heterogeneous population, able to induce T-cell tolerance by multiple mechanisms, still not completely elucidated. It is well known that MDSCs express high levels of V-domain Ig suppressor of T-cell activation (VISTA) and PD-L1, but also indoleamine 2,3-dyoxigenase (IDO), arginase, ROS, TGFβ, and IL-10 [52][53][54][55][55,56,57,58].3. Monoclonal Antibodies
3.1. Potential Targets on LSCs
The main mechanism of action of unconjugated antibodies is antibody-dependent cell-mediated cytotoxicity (ADCC). After the formation of an immunological synapse with the target cell, NK cells trigger a cytolytic response through the exocytosis of granules containing perforin and granzyme into the target cell. Furthermore, they facilitate antibody-dependent phagocytosis (ADCP) by macrophages. Their major limitation is the presence of inhibitory signals counteracting their action and the limited potency of activation signals.3.1.1. CD33
CD33 is a 67 kDa glycoprotein, a member of the siglec family (siglec-3), expressed in normal hematopoiesis from early myelo-monocytic lineage-committed progenitors to mature cells. It is also expressed in 99% of AML cells and in LSCs [56][79]. The use of unconjugated anti-CD33 is challenging due to its low membrane density and the low antibody-induced internalization, such that the first phase 3 trial (NCT0006045) with humanized IgG1 unconjugated antibody anti-CD-33, lintuzumab (HuM195) was stopped due to lack of efficacy [57][80]. Despite this, four phase I/II trials are ongoing to test a new anti-CD33 unconjugated IgG1 (BI 836858), alone or in combination with azacytidine (AZA), decitabine (DAC), or F16-IL2, in relapsed/refractory and newly diagnosed AMLs (NCT01690624, NCT03013998, NCT02032721, NCT03207191). The principal advantage of antibody–drug conjugates is the simultaneous selective attack of target cells and the on-site delivery of the potent conjugated cytotoxic agent, not usable as free drugs in conventional chemotherapy schemes for their toxicity. After the spontaneous withdrawal of the manufacturer due to the lack of survival improvement and the high induction mortality, the anti-CD33-calicamycin conjugate gemtuzumab-ozogamicin (GO) has been revived for core binding factor (CBF) AMLs and for elderly patients with AML or high-risk myelodysplastic syndrome (MDS), where it has demonstrated reduced relapse risk and survival advantage in patients with favorable and intermediate cytogenetic risk [58][81].3.1.2. CD123
CD123 is the interleukin (IL)-3 receptor alpha chain and is a type I transmembrane glycoprotein [59][83]. CD123 is present in 98% of CD34+CD38- LSCs and on blast cells but not in normal hematopoietic progenitors, making it a potential therapeutic target [14][17]. The unconjugated antibody (talacotuzumab) failed to demonstrate efficacy in AML and MDS (NCT0299860). The phase I/II trial with anti-CD123-DGN462 conjugate, alone or with AZA± VEN in MRD+ post-induction therapy AMLs, is still recruiting patients (NCT04086264).3.1.3. CLL-1
C-type lectin-receptor1 (CLL-1) is an ITIM-containing inhibitory transmembrane protein code on chromosome 12p13.31. CLL-1 is not expressed in normal HSCs, but it is present in committed progenitors and in mature peripheral myeloid cells such as monocytes, granulocytes, and dendritic cells [60][84]. Being selectively expressed in LSCs, even more than CD123, which is also present in some CD34+CD38- normal progenitors, CLL-1 could be regarded as an ideal target for anti-LSC immune therapies [19][61][22,86]. Despite CLL-1 efficiently internalizing after ligand binding, in vitro experiments with an anti-CLL-1 antibody suggested that an un-conjugated antibody cannot have anti-leukemic activity because it does not activate ADCC [19][22].3.1.4. Other Current Clinical Trials of Toxin-Conjugate Antibodies
FLT3 (FMS-like tyrosine kinase 3) is a member of the class III receptor tyrosine kinase family that is highly expressed in the blasts of both AML and ALL patients. FLT3 plays an important but not absolute role in maintaining the survival of normal HSCs, and its recurrent mutations (ITD-FLT3, TDK-FLT3) are expressed in many AML cases [62][63][89,90]. Working in conjunction with other growth factors, FLT3 promotes the proliferation and differentiation of myeloid and lymphoid cells. Transplant animal models with non-functioning and wild-type FLT3 showed that hematopoiesis is almost normal in FLT3 knockout animals, while FLT3 mutations give a significant growth advantage. These facts suggest that selective FLT3 inhibition in leukemia cells can block excessive FLT3 leukemia activation with acceptable hematopoietic side effects [63][90]. The phase I trial (NCT02864290) testing AGS62P1 anti-FLT3 antibody-amberstatin 269 in relapsed/refractory adult AMLs has been closed for lack of efficacy. NCT03957915, a phase I trial with INA03 drug-conjugate antibody-targeting transferrin receptor (CD71), is active, not recruiting, and NCT01830777, testing brentuximab-vedotin in CD30+ relapsed AMLs, is completed (results not yet available).3.2. Immune Checkpoint Inhibitors
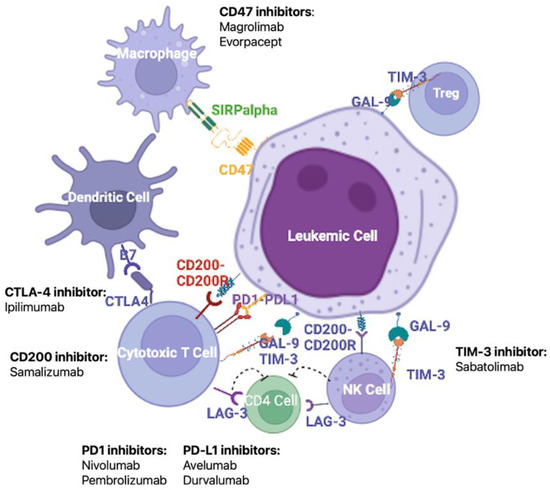
The immune system activity is closely modulated by the interaction between co-inhibitory molecules and their ligands. These co-inhibitory molecules are involved in the maintenance of immune tolerance, but in the neoplastic setting may represent one of the mechanisms employed by cancer cells to escape immune surveillance [64][65][92,93]. Many studies have reported an increased expression of co-inhibitory molecules in solid cancers, and, more recently, similar results have been observed in AML patients [66][67][94,95]. The up-regulation of co-inhibitory ligands has been associated with poor clinical outcomes in solid and hematological cancers [65][66][93,94], thus identifying a potential new class of therapy targets (Figure 12).