Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sebastian Kowalczyk and Version 3 by Jin Wang.
Atom transfer radical polymerization (ATRP) is a robust polymerization method that was developed by Dr. Jin-Shan Wang ind presented by Professor Matyjaszewski’s laboratorygroup in 1995. It was inspired by atom transfer radical addition, which was successfully used in the synthesis of low-molecular-weight compounds.
- drug delivery system
- drug nanocarrier
- micelle
- ATRP
1. Definition
Atom transfer radical polymerization (ATRP) is a robust polymerization method that was developed and presented by Professor Matyjaszewski’s group in 1995 [1][2][36,37]. It was inspired by atom transfer radical addition, which was successfully used in the synthesis of low-molecular-weight compounds [3][38]. ATRP, next to nitroxide-mediated polymerization or RAFT [4][5][39,40], is a method of CRP [6][41].
2. The Principles of ATRP
The idea of ATRP is based on reducing the concentration of radicals in the polymerization system through reversible reactions of the activation and deactivation of the active center as a result of halogen atom transfer, based on a dynamic equilibrium between working radical centers and dormant organic halides, with a relatively low homolytic dissociation activation energy of C-X bonds (Figure 1) [7][8][42,43]. Therefore, this equilibrium must be strongly shifted to the left (towards the organic halides R-X). The transfer of the halogen atom (X) from the initiator molecule or the growing polymer chain (R-X) takes place to the catalyst molecule, which is an inorganic salt, i.e., a transition metal halide with two oxidation states differing by one electron (Mt(n+)Xn), complexed with a ligand (L). For accepting a halogen, the catalyst is in a reduced form and acts as an activator (Mtn+Xn), increasing its degree of oxidation, and the process itself is an activation reaction (ka). As a result of activation, a radical (R•) is formed capable of attaching to monomer (M) molecules (initiation or propagation (kp) stage) or other reactions typical of radical polymerization. However, the participation of termination (reaction with the other radical, kt) is drastically diminished by reducing the concentration of radicals to a level several orders of magnitude lower than that of typical radical polymerization. During the operation of the radical, an appropriate number of monomer molecules are attached to the growing chain. In contrast, the number of attached monomers per one act of activation depends on many rate constants, including those that affect the ATRP equilibrium (ka, kd). The latter, in turn, depends on the structure of the organic halide and the type of catalyst, and in particular on the type of ligand whose task is to create a complex with an inorganic salt that is soluble in organic media and to give the catalyst an appropriate reduction potential [9][44]. It is worth noting that most often the role of ATRP ligands is played by polydentate nitrogen compounds, including aliphatic, cycloaliphatic, or aromatic amines [10][11][45,46]. However, other systems, for example based on phosphorus compounds, are also used [12][13][47,48]. In the next stage, the oxidized catalyst (deactivator, Mt[(n+1)+]Xn+1/L), having an additional halogen atom, reacts with one of the radicals present in the system, transferring a halide to it, which results in the formation of a dormant form of the polymerization center and the reduction of the catalyst. Due to the statistical nature of the activation and deactivation reactions, random organic halides and radicals undergo it, respectively, which, with a sufficiently fast deactivation, ensures a relatively uniform growth in all the polymer chains present in the system (linear increase in average molar mass with the degree of monomer conversion and a small dispersity in the molar masses of the obtained polymer). Copper salts were one of the first and the best-known catalytic systems used in ATRP; numerous studies have confirmed the possibility of using the halides of other metals, i.e., Fe [12][13][14][15][47,48,49,50], Ru [16][17][51,52], Ga, or Ir [18][19][53,54]. The required amount of catalyst in a normal ATRP mechanism is relatively large, as, typically, one molecule of catalyst is used per one initiation site. This has been a huge disadvantage of the method and constitutes a limitation for implementing it into industrial practice.
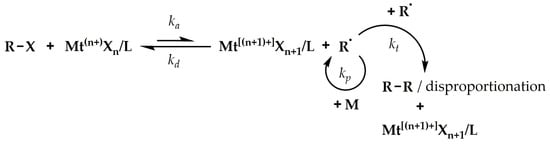
Figure 1.
General scheme of an ATRP mechanism.
In the last 15 years, numerous related methods have been developed that use much smaller amounts of catalysts. Such systems, however, require more active ligands and additional agents whose task is to regenerate the activator molecules that were irreversibly formed in the system due to some termination processes and/or the contamination of the system with oxygen. Such agents might be glucose, ascorbic acid, hydrazine, tin(II) compounds (in activators regenerated by electron transfer variant, ARGET) [20][55], typical radical initiators as sources of radicals (in initiators for continuous activator regeneration variant, ICAR) [16][21][22][23][29,51,56,57], zero covalent metals, e.g., Cu0 (in supplemental activation reducing agent variant, SARA) [24][58], or external stimuli such as electrical current/potential (in eATRP) [25][59], and ultrasound in the presence or absence of piezoelectric materials (in mechano/sonoATRP) [26][27][60,61]. The other and “greener” type of ATRP is photoinduced organocatalyzed polymerization (O-ATRP), which does not involve any metal catalyst [28][29][22,62]. These developments changed ATRP into a technique well suited to the principles of green chemistry [30][63].
Activated organic halides are used as initiators of ATRP. They can be simple compounds, such as the commonly used 2-bromobutyric acid ethyl ester, or functional initiators, i.e., those that allow the introduction of an end-group capable of chemical reactions or interactions, e.g., dye moiety [31][32][64,65], drug or proteins molecules [33][34][35][66,67,68], stimuli-sensitive molecules [36][69] or groups able to chemically react with other molecules or chains [37][70], etc. In some cases, the initiator can be covalently bonded to some surface or is able to influence the topology of macromolecules [38][39][71,72]. Therefore, ATRP allows the synthesis of linear, cyclic, and branched polymers, including star-shaped, comb, bottle-brush, and hyperbranched structures, nanogels, and polymer–drug conjugates (Figure 2).
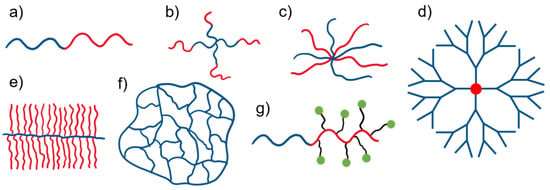
Figure 2. Representative molecular architectures synthesized via ATRP and applied in the formation of polymeric DDSs: (a) linear block copolymers, (b) regular star copolymers, (c) miktoarm star (co)polymers, (d) polymer brushes, (e) dendrimers, (f) nanogels, and (g) polymer–drug conjugate.
In the case of polymer stars, there are several synthetic strategies possible; they can be obtained via “core-first”, “arm-first”, and “coupling-onto” methods utilizing ATRP. While the “core-first” method uses a multifunctional initiator with a specified number of initiating species, which defines the number of arms, the “arm-first” method relies on ready, halogen functionalized polymer chains that are activated in the presence of the crosslinking agent, so core formation takes place in situ. It does not allow us to control the number of arms in a very good manner, however; it enables the synthesis of stars with structurally different arms (miktoarm). Polymer grafts (combs and bottle-brushes) might be synthesized by the polymerization of macromonomers (obtained by ATRP or in the other way), by the initiation of ATRP with a formed polymer chain comprising initiating species along it, or by coupling the side chains to the main chain consisting of appropriate chemical groups in monomeric units, when the side and main chains may be obtained via ATRP as well [40][73]. There have been successful attempts at star-brush molecule (“hairy” stars) synthesis as well [41][74].
It is essential that the halogen atom is formally present at the other end of the polymer chain after polymerization. This provides a vast opportunity to exchange the atom into the other, more reactive group, enabling further reactions, i.e., linking specific molecules to that chain-end or coupling the macromolecule with another one. The example of a halogen-to-azide group exchange best demonstrates this, which then opens the way to a variety of coupling possibilities with alkyne moieties, called “click” chemistry [42][75].
On the other hand, the residual halogen atom at the end of the macromolecule can be used as a macroinitiator for the synthesis of block copolymers, thus depriving ionic polymerizations of the monopoly for obtaining well-defined segment systems [43][44][76,77]. This, in turn, enables the design of materials with specific morphology obtained due to the self-assembly of block copolymers [45][78]. These include hydrophobic copolymers capable of forming ordered nanostructures from a polymer melt, e.g., lamellae, gyroids, hexagonally packed cylinders, or spheres. It is worth noting that the type of nanostructure can be influenced by the basic structural parameters included in the phase diagrams, i.e., the interaction parameter, degree of polymerization, and volume fractions of components as a predominance factor. However, ATRP allows us to control other subtle parameters, e.g., the topology or dispersion of blocks [46][47][79,80], which can change phase boundaries and stabilize phases considered to be thermodynamically metastable, e.g., hexagonal perforated lamellae [48][81].
ATRP also enables the synthesis of hydrophobic segments for amphiphilic block copolymers or complete amphiphilic block copolymers and double hydrophilic block copolymers [49][50][51][52][82,83,84,85]. Amphiphilic materials can self-assemble in aqueous systems to form vesicles (liposomes) or micelles (spherical, cylindrical, hexagonally packed, or lamellar ‘‘neat’’ micelles in parallel arrangement) [53][86], which are often used in DDSs, for instance. In some cases of doubly hydrophilic copolymers, one of the segments is a polyelectrolyte (often a polyanion), which can interact with particles of inorganic salts, being the basis for artificial biomineralization processes. Among the monomers giving hydrophilic segments in ATRP, acrylamide, 2-hydroxyethyl methacrylate (HEMA), or poly(ethylene glycol) methacrylates should be mentioned [54][87]. However, polyelectrolytes usually have to be created using hydrophobic precursors, e.g., tert-butyl acrylate (tBuA), which, after polymerization, are hydrolyzed to poly(acrylic acid) (PAA) [55][88]. The direct use of acrylic acid in polymerization causes its reaction with a deactivator, i.e., copper(II) salts. This shows that although ATRP is quite robust to reaction conditions, this method has some limitations. The main challenge of this method is the limited range of monomers that can be used compared to that in simple radical polymerization. It is required that these monomers have high resonance stabilization. Hence styrenes, acrylates, methacrylates, or acrylonitrile are most often used. At the same time, the well-controlled ATRP of ethylene, butadiene, or vinyl acetate is practically impossible. It should be noted, however, that ATRP allows us to obtain the large family of amphiphilic copolymers by using hydrophilic macroinitiators, such as poly(ethylene glycol) (PEG), and naturally occurring polysaccharides. The typical synthetic approach involves the transformation of the hydroxyl end-group of the hydrophilic reagent with 2-bromoisobutyryl bromide (BIBB) to form an ATRP macroinitiator (Figure 3) [49][82].
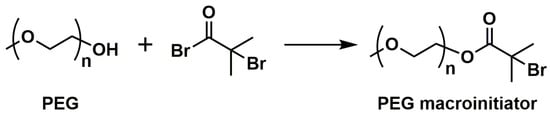
Figure 3.
A typical synthesis of PEG macroinitiator of ATRP for amphiphilic block copolymers.