Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Baptiste Panthu.
Lactic acidosis, a hallmark of solid tumour microenvironment, originates from lactate hyperproduction and its co-secretion with protons by cancer cells displaying the Warburg effect. Long considered a side effect of cancer metabolism, lactic acidosis is now known to play a major role in tumour physiology, aggressiveness and treatment efficiency.
- lactic acidosis
- glucose deprivation
- tumour heterogeneity
1. Introduction
Lactic acidosis was shown to impact numerous aspects of energy metabolism. Here focus on nutrient import, glycolysis, the tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OxPhos), and pathways generating reduced coenzymes (Figure 1).
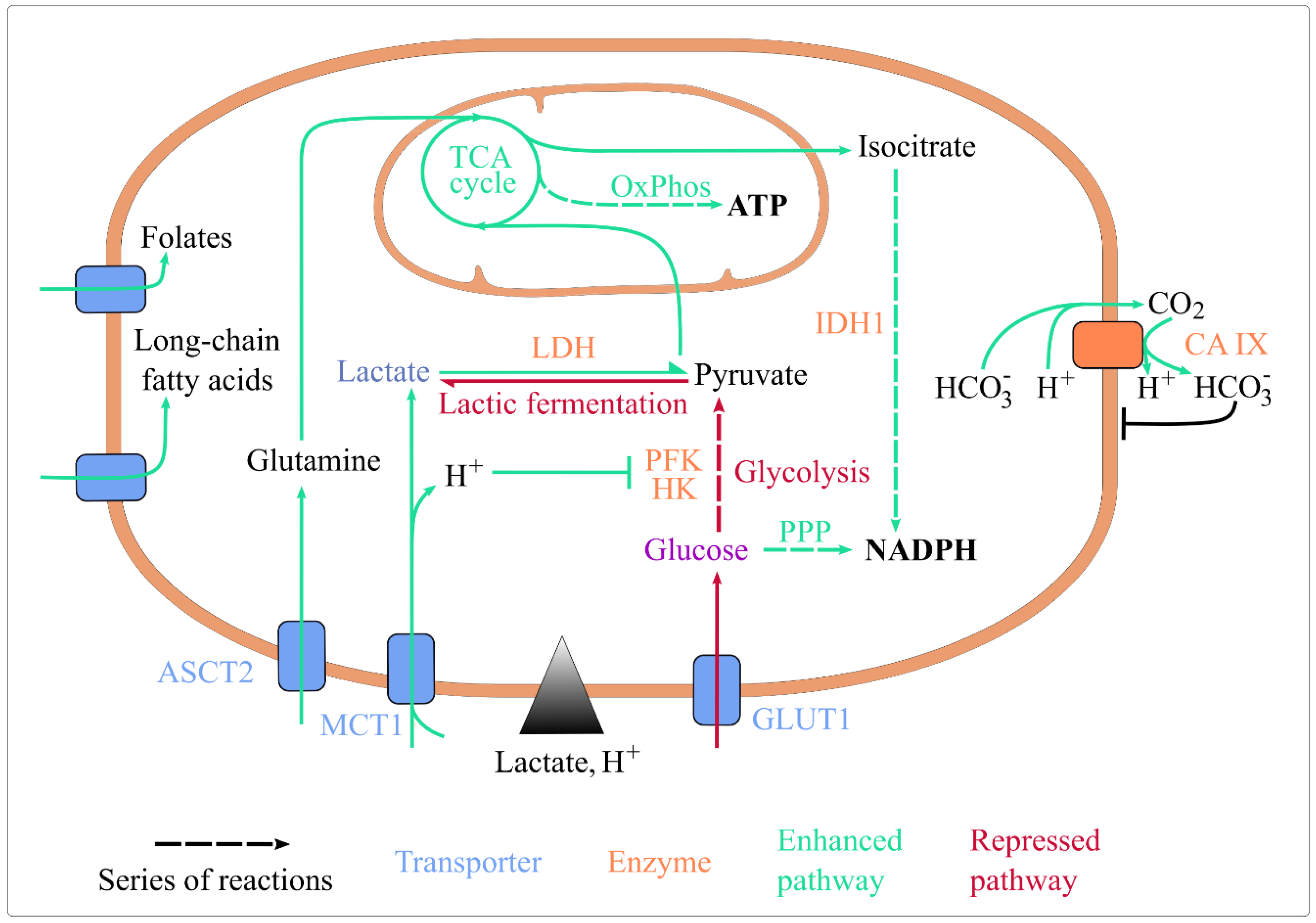
Figure 1. Lactic acidosis rewires energy metabolism and maintains cellular homeostasis. Lactic acidosis enhances the uptake of folate, long-chain fatty acids, glutamine, and lactate. It represses glucose import, glycolysis (by inhibiting HK and PFK, its rate-limiting enzymes) and lactic fermentation. It enhances lactate conversion to pyruvate, routing of pyruvate and glutamine towards the TCA cycle, ATP generation by OxPhos, and coenzyme reduction by IDH1 and the oxidative PPP. It also upregulates CA IX expression, which basifies intracellular pH. Abbreviations: ASCT2: Alanine, Serine, Cysteine Transporter 2; CA IX: carbonate anhydrase 1; GLUT1: glucose transporter 1; HK: hexokinase; IDH1: isocitrate dehydrogenase 1; MCT1: monocarboxylate transporter 1; OxPhos: oxidative phosphorylation; PFK1: phosphofructokinase 1; PPP: pentose phosphate pathway; TCA: tricarboxylic acid.
2. Lactic Acidosis and Exchanges at the Plasma Membrane
In glucose deprivation, the capacity of cancer cells to uptake and metabolise alternative nutrients is key to their survival [56][1]. Extracellular acidosis and lactosis were shown to increase such capacity.
2.1. Acidosis Sustains the Activity of Proton-Nutrient Symporters
Extracellular acidosis has a direct impact on exchanges at the plasma membrane [57][2]. In healthy tissues, protons are more concentrated inside the cell than outside. In tumours, the contrary is true [58,59][3][4]. Extracellular acidosis inverts the transmembrane proton gradient in tumour cells, which may positively impact proton-nutrient symports. Of interest, lactate is imported in cancer cells via the monocarboxylate transporters (MCTs) [51][5]. Since MCTs co-transport lactate with a proton, lactate import should be sensitive to the proton gradient and facilitated under acidosis. This mechanism is expected to explain why cancer cells respond differently to lactosis and lactic acidosis [3][6], since a rise in extracellular lactate only increases intracellular lactate levels in acidic conditions [60][7]. The co-transport of extracellular lactate and protons probably underlies the synergy of their effects on intracellular metabolism (Figure 1).
Of note, protons are also co-imported with several other nutrients, such as Fe2+, folates, amino acids and peptides. Brown & Ganapathy suggested that acidosis may affect their uptake (Figure 1), but this hypothesis remains to be confirmed [61][8].
2.2. Lactate and Acidosis Indirectly Enhance Nutrient Uptake
Extracellular lactic acidosis indirectly promotes the uptake of several nutrients (Figure 1). The import of lactate itself is increased in lactic acidosis, in part due to MCT1 overexpression [11][9], which is a response to an extracellular lactate signal [62][10] that is potentiated by extracellular acidity [12][11]. Extracellular lactate induces MCT1 and MCT4 via the G-protein-coupled receptor 81 (GPR81) transduction pathway [62][10]. In acidosis, extracellular lactate can also induce GPR81 expression [12,13][11][12]. Extracellular lactate signal enhancing MCT-mediated lactate import is necessary to cancer cell survival in absence of glucose, glutamine and pyruvate [62][10]. This supports the idea that ‘lactate induces its own metabolism’, which doesn’t exclude other regulations of MCT expression [63][13].
Glutamine uptake is increased by extracellular lactate or acidosis, as both conditions increase the expression of the glutamine transporter ASCT2 (alanine, serine and cysteine transporter 2) [14,64][14][15]. Fatty acid uptake is enhanced by acidosis [65][16], and folate import is intensified by 10 mM extracellular lactic acid [15][17].
Finally, and importantly, lactic acidosis seems to minimise glucose uptake, but not in all cell lines and cancers. Lactic acidosis decreases glucose uptake in various cell lines [5][18], as lactate in lung cancer cell lines [16][19]. On the opposite, 10 mM lactate has no effect on glucose uptake in the T47D breast cancer cell line [15][17]. The expression of the glucose transporters GLUT1 and GLUT4 are decreased by 2 mM lactate and acidosis in lung and breast cancer cell lines [17][20], and by acidosis in cervix, pharynx and colon cancer cell lines [64][15] but not in lung cancer cell lines [18][21].
2.3. Lactic Acidosis and pH Homeostasis
Cell exposure to lactic acidosis is associated with a drop in intracellular pH from 7.3 to ~6.9 [5][18]. Behind this acidification, several probable effects may be discerned. On the one hand, as discussed earlier, acidosis enhances proton-nutrient co-import, which could contribute to cellular acidification. On the other hand, lactate as a signal can mitigate the drop in pH by favouring alkalinization. A level of 10 mM extracellular lactate induces Carbonic Anhydrase IX (CA IX) [19][22], a transmembrane enzyme supporting proton export and a key regulator of cell pH [66][23] (Figure 1).
3. Lactic Acidosis, Glycolysis, and Lactic Fermentation
Unlike cells showing the Warburg effect, in which glycolysis and lactate dehydrogenase (LDH)-catalysed lactic fermentation are known to be hyperactive, cells exposed to lactic acidosis show a reduction in these pathways’ activity.
In glucose abundance, acidosis and lactic acidosis lower glucose consumption and lactate secretion [4[18][24][25],5,20], which indicates that glycolysis and lactic fermentation are downregulated. More interestingly, lactic acidosis decreases cancer cell dependency on glucose catabolism [1][26]. Thus, in glucose sufficiency, lactic acidosis minimises glucose catabolism activity and its importance in cell survival (Figure 1).
Glycolysis and lactic fermentation are likely downregulated at the level of both gene expression and enzyme activity. The expression of glycolysis enzymes is reduced by lactic acidosis in breast cancer cell lines [10][27], and by extracellular lactate in lung cancer cell lines [16][19], but it is maintained by extracellular lactate in breast cancer cell lines [21,22][28][29]. The activity of glycolysis enzymes, especially the rate-limiting hexokinase and phosphofructokinase [67][30], is directly decreased by intracellular acidification [4][24] (Figure 1). In line, intracellular acidification has been predicted in silico to hinder the Warburg effect [68][31]. Intracellular lactate accumulation, in parallel, directly inhibits lactic fermentation [5][18] (Figure 1). The interconversion of lactate and pyruvate through LDH follows the mass action law, therefore a rise in lactate concentration inhibits its production from pyruvate and favours the reverse reaction. This thermodynamic effect leads to a complete stop of lactic fermentation at ~25 mM intracellular lactate [5][18]. This concentration is within the range resulting from lactic acidosis.
4. Lactic Acidosis and Mitochondrial Catabolism
4.1. Lactic Acidosis Intensifies Mitochondrial Catabolism
Lactic acidosis enhances mitochondrial metabolic activity, in particular the TCA cycle and OxPhos. Both lactic acidosis and lactosis enhance mitochondrial biogenesis [23,24][32][33] and the expression of the enzymes of the TCA cycle and OxPhos [11[9][34],25], which potentiates mitochondrial catabolism and ATP production. The reactivation of those pathways allows the maintenance of the cellular ATP concentration in glucose deprivation and increases resistance to starvation [11][9].
4.2. Lactic Acidosis Shapes TCA Cycle Alternative Fueling
In addition to glucose-derived pyruvate, the TCA cycle can be supplied with various substrates. This flexibility is particularly true of cancer cells [69][35]. In challenging nutritional contexts such as glucose deprivation, the TCA cycle of cancer cells can be sustained by alternative nutrients. Lactate and glutamine are its main substrate suppliers after glucose [70][36]. The use of both is promoted by lactic acidosis.
The pyruvate generated from lactate can directly sustain the TCA cycle [26,27,28,29,30,31,71][37][38][39][40][41][42][43] (Figure 1). In line, extracellular lactate increases the mitochondrial membrane potential, and hence ATP production efficiency in OxPhos [21[28][44],72], and could even be necessary to pro-tumoural cell proliferation [32][45]. In more detail, the routing of lactate to mitochondria is debated. In the classical view, lactate is converted to pyruvate in the cytosol, then pyruvate is shuttled to mitochondria [73,74][46][47]. In addition to this classical way, Brooks et al. proposed an alternative model in which lactate would be shuttled to mitochondria via the mitochondrial lactate oxidation complex (mLOC), that includes MCT1 [75][48]. The controversy raised by this model has been well-reviewed in [76[49][50],77], that summarized the evidence for and against it in non-cancer cells. In cancer cells supplied with sufficient glucose, lactate’s contribution to the TCA cycle over glucose remains under debate: some studies suggest that lactate shuttled to mitochondria is preferred [71][43], while others question this [78][51]. Either way, under glucose deprivation, hypothesising that lactate’s contribution to the TCA cycle is of significant importance.
Glutamine is a major nutrient for cancer cells. It undergoes oxidative glutaminolysis in mitochondria, where it is processed by glutaminase 1 or 2 (GLS1/2) and then glutamate dehydrogenase 1 (GDH1) to sustain the TCA cycle. Lactic acidosis [20][25], acidosis [20[15][25],64], and lactate [14] upregulate GLS1 and GLS2 and stimulate oxidative glutaminolysis. Lactic acidosis, however, doesn’t necessarily promote glutamine consumption compared to lactosis [23][32]. In summary, either extracellular lactate, acidosis or lactic acidosis enhance glutamine utilisation by inducing glutaminase expression (Figure 1).
5. Lactic Acidosis and Redox Homeostasis
Cell survival requires redox homeostasis, i.e., controlled levels of reactive oxygen species (ROS) and redox coenzymes. The former lead to cell death when they accumulate, and the latter support the entire metabolism and cellular antioxidant defences.
Particularly, a high NADPH/NADP+ ratio kinetically favours anabolic reactions and helps keep ROS levels low. In cancer cells this ratio is abnormally high and sustains hyperactive anabolism [70][36]. High NADPH levels are supported by the oxidation of nutrients, such as lactate and glutamine via the TCA cycle and then oxidation of glutamine- and lactate-derived malate and isocitrate by the malic enzyme 1 (ME1) and Isocitrate Dehydrogenase 1 (IDH1), and mainly glucose via the pentose phosphate pathway (PPP). Redox homeostasis in cancer cells is therefore particularly sensitive to nutritional stress such as glucose deprivation.
In this condition, lactic acidosis helps stabilise the NADPH/NADP+ ratio at ~50% of its level in glucose sufficiency [3][6]. Likely, the gatekeepers of the NADPH/NADP+ ratio in glucose abundance have reduced efficiency under glucose deprivation and lactic acidosis, whereas new control mechanisms gain importance. On the one hand, glutamine use via ME1 is not necessary to the maintenance of the NADPH/NADP+ ratio under lactic acidosis [33][52]. Glutamine would indeed be completely degraded in mitochondrial catabolism instead of sustaining ME1 activity [20][25]. On the other hand, the glucose directed away from glycolysis towards the PPP would prevail more in NADPH/NADP+ maintenance under lactic acidosis. Lactic acidosis [20][25] and acidosis [34][53] respectively increase the expression and activity of glucose-6-phosphate dehydrogenase (G6PD), the first enzyme of the PPP, and lactic acidosis makes G6PD activity necessary to NADPH/NADP+ ratio maintenance and cell survival [20][25] in glucose sufficiency. However in glucose deprivation, the PPP alone cannot maintain redox balance [33][52]. Alternatively, lactate would become a key player in NADPH/NADP+ ratio maintenance, via the TCA cycle [35][54], and IDH1 [33][52].
Whether, in glucose abundance, such reprogramming strengthens cell defences against ROS level increase is uncertain. Acidosis increases ROS levels [35][54] and cell sensitivity to oxidative stress, but cell adaptation to acidosis decreases them [34][53]. Lactate import through MCT1 is key to maintain low ROS levels [79][55]. Lactic acidosis was found to either increase ROS levels, as does acidosis [20][25], or to rescue acidosis’ negative effect [35][54]. At any rate, in glucose deprivation, lactic acidosis mainly prevents increased ROS levels by providing IDH1 with its substrate [33][52].
A high NADH/NAD+ ratio supports ATP production. Lactic acidosis impact on the NADH/NAD+ ratio has not been directly investigated. However lactate use by the TCA cycle increases the NADH/NAD+ ratio in glucose deprivation [30][41]. This increase could contribute to the inhibition of glycolysis by lactic acidosis: a high NADH/NAD+ ratio would inhibit glycolysis according to the mass action law. Yet this hypothesis remains to be tested.
6. Summary
In the energy metabolism of cancer cells, acidosis and extracellular lactate act as enzymatic inhibitors, and lactate as a signal and a nutrient. They mostly curb glycolysis and lactic fermentation and enhance the TCA cycle and OxPhos (Figure 1). Acidification and lactate accumulation in the tumour microenvironment would promote and sustain an oxidative phenotype, which is fitter than the fermenting phenotype in glucose deprivation, an adverse nutritional context that is common in tumours.
References
- Papalazarou, V.; Maddocks, O.D.K. Supply and demand: Cellular nutrient uptake and exchange in cancer. Mol. Cell 2021, 81, 3731–3748.
- Elingaard-Larsen, L.O.; Rolver, M.G.; Sørensen, E.E.; Pedersen, S.F. How Reciprocal Interactions between the Tumor Microenvironment and Ion Transport Proteins Drive Cancer Progression. In From Malignant Transformation to Metastasis; Stock, C., Pardo, L.A., Eds.; Reviews of Physiology, Biochemistry and Pharmacology; Springer: Cham, Switzerland, 2020; Volume 182, pp. 1–38.
- Gillies, R.J.; Raghunand, N.; Karczmar, G.S.; Bhujwalla, Z.M. MRI of the tumor microenvironment. J. Magn. Reson. Imaging 2002, 16, 430–450.
- Gallagher, F.A.; Kettunen, M.I.; Day, S.E.; Hu, D.-E.; Ardenkjær-Larsen, J.H.; Zandt, R.I.; Jensen, P.R.; Karlsson, M.; Golman, K.; Lerche, M.H.; et al. Magnetic resonance imaging of pH in vivo using hyperpolarized 13C-labelled bicarbonate. Nature 2008, 453, 940–943.
- Sonveaux, P.; Végran, F.; Schroeder, T.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Investig. 2008, 118, 3930–3942.
- Wu, H.; Ding, Z.; Hu, D.; Sun, F.; Dai, C.; Xie, J.; Hu, X. Central role of lactic acidosis in cancer cell resistance to glucose deprivation-induced cell death. J. Pathol. 2012, 227, 189–199.
- Hu, X.; Chao, M.; Wu, H. Central role of lactate and proton in cancer cell resistance to glucose deprivation and its clinical translation. Signal Transduct. Target Ther. 2017, 2, 16047.
- Brown, T.P.; Ganapathy, V. Lactate/GPR81 signaling and proton motive force in cancer: Role in angiogenesis, immune escape, nutrition, and Warburg phenomenon. Pharmacol. Ther. 2020, 206, 107451.
- Duan, K.; Liu, Z.J.; Hu, S.Q.; Liu, Z.-J.; Hu, S.-Q.; Huo, H.-Y.; Xu, Z.-R.; Ruan, J.-F.; Sun, Y.; Dai, L.-P.; et al. Lactic acid induces lactate transport and glycolysis/OXPHOS interconversion in glioblastoma. Biochem. Biophys. Res. Commun. 2018, 503, 888–894.
- Roland, C.L.; Arumugam, T.; Deng, D.; Liu, S.H.; Philip, B.; Gomez, S.; Burns, W.R.; Ramachandran, V.; Wang, H.; Cruz-Monserrate, Z.; et al. Cell surface lactate receptor GPR81 is crucial for cancer cell survival. Cancer Res. 2014, 74, 5301–5310.
- Li, X.; Zhang, Z.; Zhang, Y.; Cao, Y.; Wei, H.; Wu, Z. Upregulation of lactate-inducible snail protein suppresses oncogene-mediated senescence through p16INK4a inactivation. J. Exp. Clin. Cancer Res. 2018, 37, 39.
- Xie, Q.; Zhu, Z.; He, Y.; Zhang, Z.; Zhang, Y.; Wang, Y.; Luo, J.; Peng, T.; Cheng, F.; Gao, J.; et al. A lactate-induced Snail/STAT3 pathway drives GPR81 expression in lung cancer cells. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2020, 1866, 165576.
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2020, 33, 48–66.
- Pérez-Escuredo, J.; Dadhich, R.K.; Dhup, S.; Cacace, A.; Van Hée, V.F.; De Saedeleer, C.J.; Sboarina, M.; Rodriguez, F.; Fontenille, M.-J.; Brisson, L.; et al. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 2015, 15, 72–83.
- Corbet, C.; Draoui, N.; Polet, F.; Pinto, A.; Drozak, X.; Riant, O.; Feron, O. The SIRT1/HIF2α axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014, 74, 5507–5519.
- Corbet, C.; Bastien, E.; de Jesus, J.P.S.; Dierge, E.; Martherus, R.; Linden, C.V.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020, 11, 454.
- Guedes, M.; Araújo, J.R.; Correia-Branco, A.; Gregório, I.; Martel, F.; Keating, E. Modulation of the uptake of critical nutrients by breast cancer cells by lactate: Impact on cell survival, proliferation and migration. Exp. Cell Res. 2016, 341, 111–122.
- Xie, J.; Wu, H.; Dai, C.; Pan, Q.; Ding, Z.; Hu, D.; Ji, B.; Luo, Y.; Hu, X. Beyond Warburg effect—Dual metabolic nature of cancer cells. Sci. Rep. 2015, 4, 4927.
- Jiang, J.; Huang, D.; Jiang, Y.; Hou, J.; Tian, M.; Li, J.; Sun, L.; Zhang, Y.; Zhang, T.; Li, Z.; et al. Lactate Modulates Cellular Metabolism through Histone Lactylation-Mediated Gene Expression in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 647559.
- Prado-Garcia, H.; Campa-Higareda, A.; Romero-Garcia, S. Lactic Acidosis in the Presence of Glucose Diminishes Warburg Effect in Lung Adenocarcinoma Cells. Front. Oncol. 2020, 10, 807.
- Giatromanolaki, A.; Liousia, M.; Arelaki, S.; Kalamida, D.; Pouliliou, S.; Mitrakas, A.; Tsolou, A.; Sivridis, E.; Koukourakis, M. Differential effect of hypoxia and acidity on lung cancer cell and fibroblast metabolism. Biochem. Cell Biol. 2017, 95, 428–436.
- Panisova, E.; Kery, M.; Sedlakova, O.; Brisson, L.; Debreova, M.; Sboarina, M.; Sonveaux, P.; Pastorekova, S.; Svastova, E. Lactate stimulates CA IX expression in normoxic cancer cells. Oncotarget 2017, 8, 77819–77835.
- Queen, A.; Bhutto, H.N.; Yousuf, M.; Syed, M.A.; Hassan, I. Carbonic anhydrase IX: A tumor acidification switch in heterogeneity and chemokine regulation. Semin. Cancer Biol. 2022, 86, 899–913.
- Gao, J.; Guo, Z.; Cheng, J.; Sun, B.; Yang, J.; Li, H.; Wu, S.; Dong, F.; Yan, X. Differential metabolic responses in breast cancer cell lines to acidosis and lactic acidosis revealed by stable isotope assisted metabolomics. Sci. Rep. 2020, 10, 21967.
- Lamonte, G.; Tang, X.; Chen, J.L.-Y.; Wu, J.; Ding, C.-K.C.; Keenan, M.M.; Sangokoya, C.; Kung, H.-N.; Ilkayeva, O.; Boros, L.G.; et al. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013, 1, 23.
- Barnes, E.M.E.; Xu, Y.; Benito, A.; Herendi, L.; Siskos, A.P.; Aboagye, E.O.; Nijhuis, A.; Keun, H.C. Lactic acidosis induces resistance to the pan-Akt inhibitor uprosertib in colon cancer cells. Br. J. Cancer 2020, 122, 1298–1308.
- Chen, J.L.-Y.; Lucas, J.E.; Schroeder, T.; Mori, S.; Wu, J.; Nevins, J.; Dewhirst, M.; West, M.; Chi, J.-T. The genomic analysis of lactic acidosis and acidosis response in human cancers. PLoS Genet. 2008, 4, e1000293.
- Jin, L.; Guo, Y.; Chen, J.; Wen, Z.; Jiang, Y.; Qian, J. Lactate receptor HCAR1 regulates cell growth, metastasis and maintenance of cancer-specific energy metabolism in breast cancer cells. Mol. Med. Rep. 2022, 26, 268.
- Ishihara, S.; Hata, K.; Hirose, K.; Okui, T.; Toyosawa, S.; Uzawa, N.; Nishimura, R.; Yoneda, T. The lactate sensor GPR81 regulates glycolysis and tumor growth of breast cancer. Sci. Rep. 2022, 12, 6261.
- Xie, J.; Dai, C.; Hu, X. Evidence That Does Not Support Pyruvate Kinase M2 (PKM2)-catalyzed Reaction as a Rate-limiting Step in Cancer Cell Glycolysis. J. Biol. Chem. 2016, 291, 8987–8999.
- Persi, E.; Duran-Frigola, M.; Damaghi, M.; Roush, W.R.; Aloy, P.; Cleveland, J.L.; Gillies, R.J.; Ruppin, E. Systems analysis of intracellular pH vulnerabilities for cancer therapy. Nat. Commun. 2018, 9, 2997.
- Romero-Garcia, S.; Prado-Garcia, H.; Valencia-Camargo, A.D.; Alvarez-Pulido, A. Lactic Acidosis Promotes Mitochondrial Biogenesis in Lung Adenocarcinoma Cells, Supporting Proliferation Under Normoxia or Survival Under Hypoxia. Front. Oncol. 2019, 9, 1053.
- Longhitano, L.; Vicario, N.; Tibullo, D.; Giallongo, C.; Broggi, G.; Caltabiano, R.; Barbagallo, G.M.V.; Altieri, R.; Baghini, M.; Di Rosa, M.; et al. Lactate Induces the Expressions of MCT1 and HCAR1 to Promote Tumor Growth and Progression in Glioblastoma. Front. Oncol. 2022, 12, 871798.
- Longhitano, L.; Giallongo, S.; Orlando, L.; Broggi, G.; Longo, A.; Russo, A.; Caltabiano, R.; Giallongo, C.; Barbagallo, I.; Di Rosa, M.; et al. Lactate Rewrites the Metabolic Reprogramming of Uveal Melanoma Cells and Induces Quiescence Phenotype. Int. J. Mol. Sci. 2022, 24, 24.
- Corbet, C.; Feron, O. Cancer cell metabolism and mitochondria: Nutrient plasticity for TCA cycle fueling. Biochim. Biophys. Acta BBA-Rev. Cancer 2017, 1868, 7–15.
- Inigo, M.; Deja, S.; Burgess, S.C. Ins and Outs of the TCA Cycle: The Central Role of Anaplerosis. Annu. Rev. Nutr. 2021, 41, 19–47.
- Park, S.; Chang, C.Y.; Safi, R.; Liu, X.; Baldi, R.; Jasper, J.S.; Anderson, G.R.; Liu, T.; Rathmell, J.C.; Dewhirst, M.W.; et al. ERRα-Regulated Lactate Metabolism Contributes to Resistance to Targeted Therapies in Breast Cancer. Cell. Rep. 2016, 15, 323–335.
- Erdem, A.; Marin, S.; Pereira-Martins, D.A.; Geugien, M.; Cunningham, A.; Pruis, M.G.; Weinhäuser, I.; Gerding, A.; Bakker, B.M.; Wierenga, A.T.J.; et al. Inhibition of the succinyl dehydrogenase complex in acute myeloid leukemia leads to a lactate-fuelled respiratory metabolic vulnerability. Nat. Commun. 2022, 13, 2013.
- Minami, N.; Tanaka, K.; Sasayama, T.; Kohmura, E.; Saya, H.; Sampetrean, O. Lactate Reprograms Energy and Lipid Metabolism in Glucose-Deprived Oxidative Glioma Stem Cells. Metabolites 2021, 11, 325.
- Montal, E.D.; Bhalla, K.; Dewi, R.E.; Ruiz, C.F.; Haley, J.A.; Ropell, A.E.; Gordon, C.; Girnun, G.D.; Haley, J.D. Inhibition of phosphoenolpyruvate carboxykinase blocks lactate utilization and impairs tumor growth in colorectal cancer. Cancer Metab. 2019, 7, 8.
- Otto, A.M.; Hintermair, J.; Janzon, C. NADH-linked metabolic plasticity of MCF-7 breast cancer cells surviving in a nutrient-deprived microenvironment. J. Cell. Biochem. 2015, 116, 822–835.
- Zhao, Y.; Li, M.; Yao, X.; Fei, Y.; Lin, Z.; Li, Z.; Cai, K.; Zhao, Y.; Luo, Z. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020, 33, 108487.
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate metabolism in human lung tumors. Cell 2017, 171, 358–371.
- Chen, L.B. Mitochondrial Membrane Potential in Living Cells. Annu. Rev. Cell Biol. 1988, 4, 155–181.
- Deng, H.; Gao, Y.; Trappetti, V.; Hertig, D.; Karatkevich, D.; Losmanova, T.; Urzi, C.; Ge, H.; Geest, G.A.; Bruggmann, R.; et al. Targeting lactate dehydrogenase B-dependent mitochondrial metabolism affects tumor initiating cells and inhibits tumorigenesis of non-small cell lung cancer by inducing mtDNA damage. Cell. Mol. Life Sci. 2022, 79, 445.
- Herzig, S.; Raemy, E.; Montessuit, S.; Veuthey, J.-L.; Zamboni, N.; Westermann, B.; Kunji, E.R.S.; Martinou, J.-C. Identification and functional expression of the mitochondrial pyruvate carrier. Science 2012, 337, 93–96.
- Bricker, D.K.; Taylor, E.B.; Schell, J.C.; Orsak, T.; Boutron, A.; Chen, Y.-C.; Cox, J.E.; Cardon, C.M.; Van Vranken, J.G.; Dephoure, N.; et al. A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila, and Humans. Science 2012, 337, 96–100.
- Hashimoto, T.; Hussien, R.; Brooks, G.A. Colocalization of MCT1, CD147, and LDH in mitochondrial inner membrane of L6 muscle cells: Evidence of a mitochondrial lactate oxidation complex. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1237–E1244.
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785.
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T. Lactate metabolism in human health and disease. Signal Transduct. Target Ther. 2022, 7, 305.
- Ying, M.; Guo, C.; Hu, X. The quantitative relationship between isotopic and net contributions of lactate and glucose to the tricarboxylic acid (TCA) cycle. J. Biol. Chem. 2019, 294, 9615–9630.
- Ying, M.; You, D.; Zhu, X.; Cai, L.; Zeng, S.; Hu, X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021, 46, 102065.
- Chen, S.; Ning, B.; Song, J.; Yang, Z.; Zhou, L.; Chen, Z.; Mao, L.; Liu, H.; Wang, Q.; He, S.; et al. Enhanced pentose phosphate pathway activity promotes pancreatic ductal adenocarcinoma progression via activating YAP/MMP1 axis under chronic acidosis. Int. J. Biol. Sci. 2022, 18, 2304–2316.
- Koncošová, M.; Vrzáčková, N.; Křížová, I.; Tomášová, P.; Rimpelová, S.; Dvořák, A.; Vítek, L.; Rumlová, M.; Ruml, T.; Zelenka, J. Inhibition of Mitochondrial Metabolism Leads to Selective Eradication of Cells Adapted to Acidic Microenvironment. Int. J. Mol. Sci. 2021, 22, 10790.
- Tasdogan, A.; Faubert, B.; Ramesh, V.; Ubellacker, J.M.; Shen, B.; Solmonson, A.; Murphy, M.M.; Gu, Z.; Gu, W.; Martin, M.; et al. Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 2020, 577, 115.
More