Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Rukman Awang Hamat and Version 2 by Lindsay Dong.
The production and use of antibiotics increased significantly after the Second World War due to their effectiveness against bacterial infections. However, bacterial resistance also emerged and has now become an important global issue. Those most in need are typically high-risk and include individuals who experience burns and other wounds, as well as those with pulmonary infections caused by antibiotic-resistant bacteria, such as Pseudomonas aeruginosa, Acinetobacter sp, and Staphylococcus sp. With investment to develop new antibiotics waning, finding and developing alternative therapeutic strategies to tackle this issue is imperative. One option remerging in popularity is bacteriophage (phage) therapy.
- phage therapy
- antibiotics resistant
- S. aureus
- biofilms
- infection
1. Introduction
Bacteriophages (phages) are found in all habitats, and their interaction with bacteria has attracted greater attention from scientists for almost a decade [1]. Bacteriophage therapy or phage therapy (PT) that consists of specific virulent bacteriophages can be used for targeting multidrug-resistant bacteria and can mimic the action of an antibacterial agent [2]. Indeed, PT has currently been used to treat patients with methicillin-resistant Staphylococcus aureus (MRSA) infections [3][4][5][6]. According to the NCBI database, a total of 69 genomes of virulent staphylophages have been deposited, with 26 and 43 genomes belonging to the family of Podoviridae and Myoviridae phages, respectively [7]. The high efficiency of staphylophages in the therapeutic cocktail is attributed to its broad host range and high lytic capabilities [8].
Although various successful investigations have explored using singular or multiple phage types (phage cocktails), a definitive answer on their safety and efficacy as part of standard clinical care is still unknown [9].
2. Phage Therapy for Resistant Staphylococcus aureus Infections
2.1. Staphylococci
Staphylococci are frequently isolated as the causal agent of bacterial infections in humans, and its management remains challenging due to the advent of multidrug-resistant strains [10]. It commonly causes surgical wound infections and pneumonia and is the second most common cause of blood infections [11]. Importantly, S. aureus has been extremely efficient at developing resistance to the most recent antibiotic classes. Although methicillin-resistant S. aureus (MRSA) was quite rare between the 1960s and 1980s, the problem escalated in the mid-1990s when particular ‘epidemic’ strains became established in hospitals throughout the UK, which were caused by SCCmec type 1. Several antibiotics targeting methicillin-resistant S. aureus (MRSA) have been developed and approved for use, including linezolid, daptomycin, tigecycline, ceftobiprole, telavancin, ceftaroline, dalbavancin, oritavancin, and tedizolid [12]. However, they have reduced efficacy when staphylococci establish as biofilms, typically seen with device-associated infections and endocarditis [13].
2.2. Genetic Structure of Staphylococcus aureus
The S. aureus genome consists of a circular double-stranded chromosome as well as several plasmids. The number of complete and draft genomes has grown exponentially, with hundreds now available in the NCBI repository [14][15]. Upon examination, conservational similarities and differences are seen between methicillin-sensitive S. aureus (MSSA) and MRSA. For example, the genomic core (representing ~75% of the entire genome) is extremely conserved between both MRSA and MSSA isolates and contains genes associated with housekeeping and other metabolic functions [16]. In addition to the core genome, accessory genomes can be obtained from other bacterial species by lateral gene transfer [17]. This domain accounts for ~25% of the entire genome and consists of mobile genetic elements such as virulence factors, chromosome cassettes, genomic plasmids, transposons, and antibiotic resistance dynamics [18].2.3. Staphylococcal Cassette Chromosome (SCC)
As an important basic mobile genetic element, the SCC serves as the motor vehicle for gene substitution among Staphylococcus species. It typically contains the mec gene (although not always present), which codes for methicillin resistance, regulatory genes, as well as the cassette chromosome recombinases (ccr), which enables the integration and cutting of SCCmec [19]. SCCmec typing enables structural determination of SCCmec and can assist in inferring the origins of particular MRSA strains [20]. Each SCCmec element integrates at the same site, which is a precise site at the 3‘ end of a unique ORF (open reading frame) of unknown function, designated orfX [21]. To date, 13 different types of SCCmec (I-XIII) have been defined according to their subparts, namely, the ccr and the mec complexes [22][23].2.4. Update on Treatment of S. aureus Infections and Its Associated Challenges
Treatment agents currently available for S. aureus infections include cloxacillin, flucloxacillin, cefazolin, ceftaroline, ceftobiprole, vancomycin, daptomycin, linezolid, teicoplanin, tigecycline, co-trimoxazole, doxycycline, clindamycin, telavancin, dalbavancin, oritavancin, and others [24]. Several antibiotic guidelines for treating S. aureus infections have been established and regularly updated according to the epidemiological data of antibiotic resistance patterns, national and global surveillance of resistance, and efficacy data of newly anti-staphylococcal agents [25]. The guideline consists of recommended antibiotics (weak and strong recommendations) and therapy regimes (dosage and duration) used for specific types of infections such as skin and soft-tissue infections (SSTIs), urinary tract infections (UTI), bone and joint infections, bacteremia, endocarditis, pneumonia, and meningitis [26]. However, infections caused by MRSA deserve special attention as common antibiotics used for MSSA are no longer effective against them, and their propensity to rapidly spread, causing an outbreak, is very common [27][28]. Topical antiseptics such as hydrogen peroxide 1% cream remain an effective option for patients with uncomplicated impetigo due to MRSA [26]. Despite new anti-MRSA agents that have been officially licensed, there is no solid evidence to establish the optimal agent(s) for specific infections caused by MRSA. Retapamulin ointment 1%, an agent of the pleuromutilin class, is still inferior to oral linezolid in a clinical trial, thus, limiting its use for this condition [29]. Ceftobiprole, dalbavancin, and tedizolid are not recommended for the SSTIs caused by MRSA due to inconclusive evidence from the clinical trials [30][31][32]. In fact, major side effects, including thrombocytopenia and optic neuropathy, have been reported in patients treated with linezolid and tedizolid [33]. Limited data from clinical trials also suggested that complicated (non-catheter related) UTIs caused by MRSA should be treated with intravenous glycopeptides such as vancomycin or teicoplanin [26]. Similarly, intravenous glycopeptides are strongly recommended for treating necrotizing pneumonia due to Panton–Valentine leucocidin (PVL)-producing MRSA strains despite no recent update about the existing guideline in the UK since 2008 [34]. As for hospital-acquired pneumonia (HAP) caused by MRSA, a similar approach to therapy is not highly recommended due to a lack of data based on clinical trials [26]. Rifampicin is used as an adjunct therapy in patients with MRSA bone or joint infections for its ability to penetrate the biofilm [34]. There has been an increasing preference for using dalbavancin among clinicians for its long half-life and convenient dosing regimen [35]. Nonetheless, recent evidence from the clinical trials fails to support the use of these drugs for this condition. Instead, intravenous glycopeptides are highly recommended with proper therapeutic drug monitoring in view of the nephrotoxic effect associated with the prolonged use of vancomycin [26][36]. A similar recommendation is proposed for MRSA bacteremia except for the use of teicoplanin. Linezolid, telavancin, and ceftaroline can be used as alternative second-line drugs if vancomycin is contraindicated [37][38], although it is not highly recommended by the recent guidelines [26].2.5. Resistance to Antimicrobial Therapy
A pathogen’s success in causing infection depends on two factors, (i) developing resistance to antibiotics and (ii) virulence factors. Resistance in S. aureus is obtained via the acquisition of mobile genetic elements (SCCmec) containing resistance genes such as mecA encoding a unique protein called penicillin-binding protein 2a (PBP2a). Its low affinity for β-lactam antibiotics thus enables it to substitute the biosynthetic functions of PBPs [39]. In addition to SCCmec, other mobile genetic elements involved in resistance acquisition include plasmids, transposons, insertion sequences, integrons, integrative-conjugative elements, and pathogenicity islands [40]. One staphylococcal conjugative plasmid (pGO1/pSK41) has been directly shown to have contributed to resistance to aminoglycosides, penicillin, trimethoprim, bleomycin, tetracycline, macrolides, lincosamide, streptogramin B, and more recently linezolid and vancomycin [41]. Furthermore, transposons, which themselves are a group of mobile genetic elements, can transfer resistance genes due to their ability to transfer between plasmids or from a DNA chromosome to a plasmid (or vice versa) [42]. Thus, antibiotic resistance emerges rapidly through the acquisition of antibiotic-resistance genes from other strains of S. aureus or even from other genera. The development of resistance due to antibiotic treatment failure results in outbreaks of multiple drug-resistant (MDR) strains, typically in hospital institutions as well as the general population [43].2.6. Role of Biofilm Formation in S. aureus Infections
Microbial populations tend to adhere to surfaces by producing extracellular polymeric substances (EPS) and forming biofilms [44]. Bacterial cells growing in a biofilm are difficult to treat by antibiotics since the EPS matrix prevents them from reaching the bacteria via physical repulsion or limited diffusion [45]. It has been estimated that of all bacterial infections, ~80% involve biofilm formation [46], and these appear to be 10–1000-fold more resistant to antibiotics compared to their planktonic counterparts [47]. The challenge to eradicate bacterial biofilms via antibiotics is well documented, with evidence suggesting some variabilities between drugs. For example, oxacillin, cefotaxime, and vancomycin have shown limited penetration, whereas amikacin and ciprofloxacin are far more effective in this function [48]. However, eradication of biofilms is very challenging, especially in clinical settings, as high rates of recurrent or chronic bacterial infections have been observed due to biofilms [49]. The development of biofilm-related S. aureus infections is always associated with medical devices or procedures used as part of patients’ management, such as indwelling catheters or implants, and chronic conditions such as chronic lung infections, chronic wounds, endocarditis, and others [49].2.7. Phage Therapy
The term “phage” is defined as a type of virus that can enter and destroy bacterial cells whose potential was appreciated early in history [50][51]. The morphological structure of a bacteriophage is unique as it consists of a head that is filled with DNA or RNA and a tail that is used for the introduction of the genome into the bacterial cells (Figure 14). It has limited receptors on eukaryotic cells; hence, it may not enter mammalian cells, making it the most promising alternative therapeutic option in humans [52][53].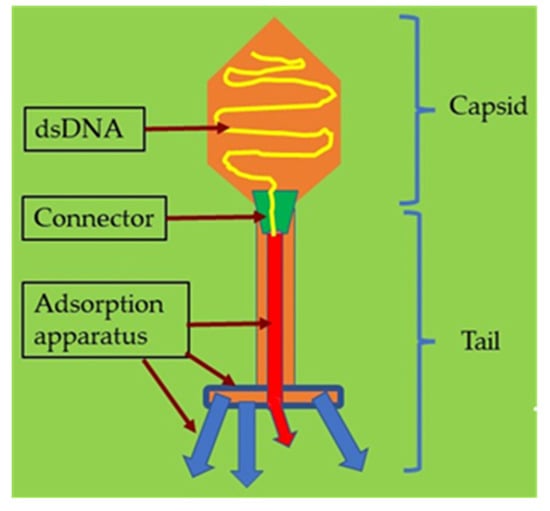
Figure 14.
Illustration of a bacteriophage with its structure and components.
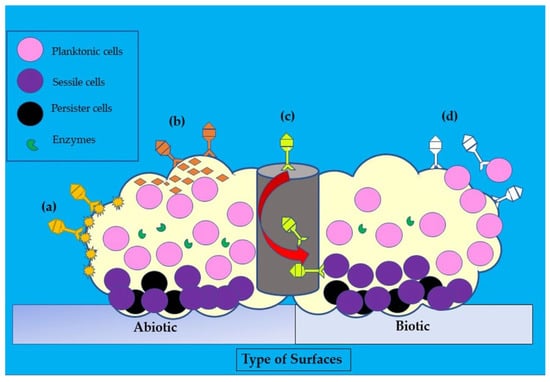
Figure 25. Illustration of the disruptive mechanisms of bacteriophages in Staphylococcus aureus biofilms on abiotic and biotic surfaces by the (a) expression of polysaccharide depolymerases which degrade extracellular matrix, (b) induction of host cells to produce exopolysaccharides (EPS)-degrading enzymes, (c) diffusion via biofilm water channels for inner layer penetration, and (d) adsorption to bacteria for biofilm penetration. The role of each bacteriophage is depicted in various colors (dark yellow, orange, yellow, and white).
Phage therapy (PT) should also be carefully assessed in patients with chronic infections caused by polymicrobial agents. For example, anti-S. aureus PT failed to completely cure chronic polymicrobial biofilm infection of a bone allograft in a sarcoma patient. The scholars explained the possibility of incomplete coverage of anti-S. aureus PT against non-staphylococci bacteria [68]. In addition, a special precaution should be implemented when PT is used when treating infections in patients with cancers. Following the introduction of bacteriophages (T4 and M13) into prostate cancer cell lines (PC-3), overexpression of integrins was observed, which may be beneficial to prostate cancer cases [69]. However, it may be detrimental to other types of cancers, such as ovarian and breast cancers. Overexpression of selected integrins (ITGAV and ITGB3) would promote the survivability of these cancer cells and their proliferation [70]. Although there is a lack of phage receptors in mammalian cells, Bichet et al. [71] demonstrated the uptake of phages by several cell lines through micropinocytosis in vitro [71]. The uptake of phages and their internalization process across the cell lines are very heterogenous, and the scholars concluded that the type of cells and phages have no roles in their specificity. Interestingly, the viability of these phages was lost due to the inactivation process [71]. This finding could further compromise the accessibility of phages to the site of infection in the tissues or organs during phage therapy. Modifications in the shape of phages may reduce the sequestration issues related to cellular uptake during phage therapy. It has been shown that the elongated-shaped phages have lower uptake rates by the cells compared with the disc-shaped phages [72]. Tolerability and safety for patients are paramount but are affected by the solution formulations phages are stored in. Formulations need to be specifically designed in order to maintain phage stability in storage, which, when poorly formulated, would impact effectiveness during treatment [73]. On a similar note, an appropriate therapeutic regime (dosage, dosing interval, and timing) is also pivotal in ensuring the efficacy of phage therapy, as clearly explained by [1].
References
- Pincus, N.B.; Reckhow, J.D.; Saleem, D.; Jammeh, M.L.; Datta, S.K.; Myles, I.A. Strain Specific Phage Treatment for Staphylococcus aureus Infection Is Influenced by Host Immunity and Site of Infection. PLoS ONE 2015, 10, e0124280.
- Breederveld, R.S. Phage therapy 2.0: Where do we stand? Lancet Infect. Dis. 2019, 19, 2–3.
- Fish, R.; Kutter, E.; Bryan, D.; Wheat, G.; Kuhl, S. Resolving Digital Staphylococcal Osteomyelitis Using Bacteriophage—A Case Report. Antibiotics 2018, 7, 87.
- Morozova, V.V.; Vlassov, V.V.; Tikunova, N.V. Applications of Bacteriophages in the Treatment of Localized Infections in Humans. Front. Microbiol. 2018, 9, 1696.
- Petrovic Fabijan, A.; Lin, R.C.Y.; Ho, J.; Maddocks, S.; Ben Zakour, N.L.; Iredell, J.R.; Khalid, A.; Venturini, C.; Chard, R.; Morales, S.; et al. Safety of bacteriophage therapy in severe Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 465–472.
- Oduor, J.M.O.; Kadija, E.; Nyachieo, A.; Mureithi, M.W.; Skurnik, M. Bioprospecting Staphylococcus Phages with Therapeutic and Bio-Control Potential. Viruses 2020, 12, 133.
- Kornienko, M.; Kuptsov, N.; Gorodnichev, R.; Bespiatykh, D.; Guliaev, A.; Letarova, M.; Kulikov, E.; Veselovsky, V.; Malakhova, M.; Letarov, A.; et al. Contribution of Podoviridae and Myoviridae bacteriophages to the effectiveness of anti-staphylococcal therapeutic cocktails. Sci. Rep. 2020, 10, 18612.
- McCallin, S.; Sarker, S.A.; Sultana, S.; Oechslin, F.; Brüssow, H. Metagenome analysis of Russian and Georgian Pyophage cocktails and a placebo-controlled safety trial of single phage versus phage cocktail in healthy Staphylococcus aureus carriers. Environ. Microbiol. 2018, 20, 3278–3293.
- Romero-Calle, D.; Guimarães Benevides, R.; Góes-Neto, A.; Billington, C. Bacteriophages as Alternatives to Antibiotics in Clinical Care. Antibiotics 2019, 8, 138.
- Taylor, T.A.; Unakal, C.G. Staphylococcus aureus. In StatPearls ; StatPearls Publishing: Tampa, FL, USA, 2022.
- Brock, T.D.; Madigan, M.T.; Martinko, J.M.; Parker, J. Brock Biology of Microorganisms, 10th ed.; Prentice-Hall: Upper Saddle River, NJ, USA, 2003.
- Kållberg, C.; Årdal, C.; Salvesen Blix, H.; Klein, E.; Martinez, E.M.; Lindbæk, M.; Outterson, K.; Røttingen, J.-A.; Laxminarayan, R. Introduction and geographic availability of new antibiotics approved between 1999 and 2014. PLoS ONE 2018, 13, e0205166.
- Molina-Manso, D.; del Prado, G.; Ortiz-Pérez, A.; Manrubia-Cobo, M.; Gómez-Barrena, E.; Cordero-Ampuero, J.; Esteban, J. In vitro susceptibility to antibiotics of staphylococci in biofilms isolated from orthopaedic infections. Int. J. Antimicrob. Agents 2013, 41, 521–523.
- Howden Benjamin, P.; Seemann, T.; Harrison Paul, F.; McEvoy Chris, R.; Stanton Jo-Ann, L.; Rand Christy, J.; Mason Chris, W.; Jensen Slade, O.; Firth, N.; Davies John, K.; et al. Complete Genome Sequence of Staphylococcus aureus Strain JKD6008, an ST239 Clone of Methicillin-Resistant Staphylococcus aureus with Intermediate-Level Vancomycin Resistance. J. Bacteriol. 2010, 192, 5848–5849.
- Pang, R.; Wu, S.; Zhang, F.; Huang, J.; Wu, H.; Zhang, J.; Li, Y.; Ding, Y.; Zhang, J.; Chen, M.; et al. The Genomic Context for the Evolution and Transmission of Community-Associated Staphylococcus aureus ST59 Through the Food Chain. Front. Microbiol. 2020, 11, 422.
- El Garch, F.; Hallin, M.; De Mendonça, R.; Denis, O.; Lefort, A.; Struelens, M.J. StaphVar-DNA microarray analysis of accessory genome elements of community-acquired methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 2009, 63, 877–885.
- Slott Jensen, M.L.; Nielsine Skov, M.; Pries Kristiansen, H.; Toft, A.; Lundgaard, H.; Gumpert, H.; Westh, H.; Holm, A.; Kolmos, H.J.; Kemp, M. Core genome multi-locus sequence typing as an essential tool in a high-cost livestock-associated meticillin-resistant Staphylococcus aureus CC398 hospital outbreak. J. Hosp. Infect. 2020, 104, 574–581.
- Balaky, S. The Effect of Antibiotics on Toxin Gene Expression in PVL-positive Staphylococcus aureus Strains. Ph.D. Thesis, Durham University, Durham, UK, 2011.
- Ioanas, M.; Lode, H. Linezolid in VAP by MRSA: A better choice? Intensive Care Med. 2004, 30, 343–346.
- Livermore, D.M. Linezolid in vitro: Mechanism and antibacterial spectrum. J. Antimicrob. Chemother. 2003, 51, ii9–ii16.
- Ito, T.; Ma Xiao, X.; Takeuchi, F.; Okuma, K.; Yuzawa, H.; Hiramatsu, K. Novel Type V Staphylococcal Cassette Chromosome mec Driven by a Novel Cassette Chromosome Recombinase, ccrC. Antimicrob. Agents Chemother. 2004, 48, 2637–2651.
- Zhang, K.; McClure, J.-A.; Elsayed, S.; Conly John, M. Novel Staphylococcal Cassette Chromosome mec Type, Tentatively Designated Type VIII, Harboring Class A mec and Type 4 ccr Gene Complexes in a Canadian Epidemic Strain of Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2009, 53, 531–540.
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18.
- Rasmussen, R.V.; Fowler Jr, V.G.; Skov, R.; Bruun, N.E. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol. 2010, 6, 43–56.
- Kavanagh, K.T.; Abusalem, S.; Calderon, L.E. View point: Gaps in the current guidelines for the prevention of Methicillin-resistant Staphylococcus aureus surgical site infections. Antimicrob. Resist. Infect. Control 2018, 7, 112.
- Brown, N.M.; Goodman, A.L.; Horner, C.; Jenkins, A.; Brown, E.M. Treatment of methicillin-resistant Staphylococcus aureus (MRSA): Updated guidelines from the UK. JAC-Antimicrob. Resist. 2021, 3, dlaa114.
- Vonberg, R.P.; Weitzel-Kage, D.; Behnke, M.; Gastmeier, P. Worldwide Outbreak Database: The largest collection of nosocomial outbreaks. Infection 2011, 39, 29–34.
- Rubin, I.M.; Hansen, T.A.; Klingenberg, A.M.; Petersen, A.M.; Worning, P.; Westh, H.; Bartels, M.D. A Sporadic Four-Year Hospital Outbreak of a ST97-IVa MRSA With Half of the Patients First Identified in the Community. Front. Microbiol. 2018, 9, 1494.
- Tanus, T.; Scangarella-Oman, N.E.; Dalessandro, M.; Li, G.; Breton, J.J.; Tomayko, J.F. A Randomized, Double-blind, Comparative Study to Assess the Safety and Efficacy of Topical Retapamulin Ointment 1% Versus Oral Linezolid in the Treatment of Secondarily Infected Traumatic Lesions and Impetigo Due to Methicillin-Resistant Staphylococcus aureus. Adv. Ski. Wound Care 2014, 27, 548–559.
- Noel, G.J.; Bush, K.; Bagchi, P.; Ianus, J.; Strauss, R.S. A Randomized, Double-Blind Trial Comparing Ceftobiprole Medocaril with Vancomycin plus Ceftazidime for the Treatment of Patients with Complicated Skin and Skin-Structure Infections. Clin. Infect. Dis. 2008, 46, 647–655.
- Boucher, H.W.; Wilcox, M.; Talbot, G.H.; Puttagunta, S.; Das, A.F.; Dunne, M.W. Once-Weekly Dalbavancin versus Daily Conventional Therapy for Skin Infection. New Engl. J. Med. 2014, 370, 2169–2179.
- Moran, G.J.; Fang, E.; Corey, G.R.; Das, A.F.; De Anda, C.; Prokocimer, P. Tedizolid for 6 days versus linezolid for 10 days for acute bacterial skin and skin-structure infections (ESTABLISH-2): A randomised, double-blind, phase 3, non-inferiority trial. Lancet Infect. Dis. 2014, 14, 696–705.
- John, J., Jr. The treatment of resistant staphylococcal infections. F1000Research 2020, 9, 150.
- Gould, F.K.; Brindle, R.; Chadwick, P.R.; Fraise, A.P.; Hill, S.; Nathwani, D.; Ridgway, G.L.; Spry, M.J.; Warren, R.E.; on behalf of the Joint Working Party of the British Society for Antimicrobial Chemotherapy, Hospital Infection Society and Infection Control Nurses Association. Guidelines (2008) for the prophylaxis and treatment of methicillin-resistant Staphylococcus aureus (MRSA) infections in the United Kingdom. J. Antimicrob. Chemother. 2009, 63, 849–861.
- Dunne Michael, W.; Puttagunta, S.; Sprenger Craig, R.; Rubino, C.; Van Wart, S.; Baldassarre, J. Extended-Duration Dosing and Distribution of Dalbavancin into Bone and Articular Tissue. Antimicrob. Agents Chemother. 2015, 59, 1849–1855.
- Filippone, E.J.; Kraft, W.K.; Farber, J.L. The Nephrotoxicity of Vancomycin. Clin. Pharmacol. Ther. 2017, 102, 459–469.
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical Practice Guidelines by the Infectious Diseases Society of America for the Treatment of Methicillin-Resistant Staphylococcus aureus Infections in Adults and Children. Clin. Infect. Dis. 2011, 52, e18–e55.
- Choo, E.J.; Chambers, H.F. Treatment of Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect. Chemother. 2016, 48, 267–273.
- Robinson, D.A.; Enright Mark, C. Evolutionary Models of the Emergence of Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2003, 47, 3926–3934.
- Rolo, J.; Worning, P.; Boye Nielsen, J.; Sobral, R.; Bowden, R.; Bouchami, O.; Damborg, P.; Guardabassi, L.; Perreten, V.; Westh, H.; et al. Evidence for the evolutionary steps leading to mecA-mediated β-lactam resistance in staphylococci. PLOS Genet. 2017, 13, e1006674.
- Cafini, F.; Romero, V.M.; Morikawa, K. Mechanisms of horizontal gene transfer. In The Rise of Virulence Antibiotic Resistance in Staphylococcus aureus; Intech Open: Rijeka, Croatia, 2017; Volume 61.
- Babakhani, S.; Oloomi, M. Transposons: The agents of antibiotic resistance in bacteria. J. Basic Microbiol. 2018, 58, 905–917.
- Pillar, C.M.; Draghi, D.C.; Sheehan, D.J.; Sahm, D.F. Prevalence of multidrug-resistant, methicillin-resistant Staphylococcus aureus in the United States: Findings of the stratified analysis of the 2004 to 2005 LEADER Surveillance Programs. Diagn. Microbiol. Infect. Dis. 2008, 60, 221–224.
- Monroe, D. Looking for Chinks in the Armor of Bacterial Biofilms. PLOS Biol. 2007, 5, e307.
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18.
- Hsu, L.-Y.; Koh, T.-H.; Singh, K.; Kang, M.-L.; Kurup, A.; Tan, B.-H. Dissemination of Multisusceptible Methicillin-Resistant Staphylococcus aureus in Singapore. J. Clin. Microbiol. 2005, 43, 2923–2925.
- Mah, T.-F.C.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39.
- Stenz, L. The GTP-Dependant Pleiotropic Repressor” CodY” Regulates Biofilm Formation in Staphylococcus aureus. Ph.D. Thesis, University of Geneva, Geneva, Switzerland, 2011.
- Lebeaux, D.; Ghigo, J.-M.; Beloin, C. Biofilm-Related Infections: Bridging the Gap between Clinical Management and Fundamental Aspects of Recalcitrance toward Antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543.
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783.
- Reza, A.; Farid, A.J.; Zamberi, S.; Amini, R.; Sajedeh, K.; Ahmad, N.; Hassan, A.; Norkhoda, S.; Morovat, T.; Iraj, P. Dynamics of bacteriophages as a promising antibiofilm agents. J. Pure Appl. Microbiol. 2014, 8, 1015–1019.
- Miernikiewicz, P.; Dąbrowska, K.; Piotrowicz, A.; Owczarek, B.; Wojas-Turek, J.; Kicielińska, J.; Rossowska, J.; Pajtasz-Piasecka, E.; Hodyra, K.; Macegoniuk, K.; et al. T4 Phage and Its Head Surface Proteins Do Not Stimulate Inflammatory Mediator Production. PLoS ONE 2013, 8, e71036.
- Van Belleghem, J.D.; Dąbrowska, K.; Vaneechoutte, M.; Barr, J.J.; Bollyky, P.L. Interactions between Bacteriophage, Bacteria, and the Mammalian Immune System. Viruses 2019, 11, 10.
- Letarov, A.V.; Kulikov, E.E. Adsorption of bacteriophages on bacterial cells. Biochem. 2017, 82, 1632–1658.
- Twort, F.W. An investigation on the nature of ultra-microscopic viruses. Acta Kravsi 1961, 189, 1241–1243.
- d’Herelle, F. On an invisible microbe antagonistic to dysentery bacilli. CR Acad. Sci. 1917, 165, 373–375.
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85.
- Międzybrodzki, R.; Borysowski, J.; Weber-Dąbrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawełczyk, Z.; Rogóż, P.; Kłak, M.; Wojtasik, E.; et al. Chapter 3—Clinical Aspects of Phage Therapy. In Advances in Virus Research; Łobocka, M., Szybalski, W., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 83, pp. 73–121.
- Chanishvili, N. Literature Review of the Practical Application of Bacteriophage Research; Nova Science Publishers, Incorporated: New York, NY, USA, 2012.
- Chan, B.K.; Abedon, S.T. Chapter 1—Phage Therapy Pharmacology: Phage Cocktails. In Advances in Applied Microbiology; Laskin, A.I., Sariaslani, S., Gadd, G.M., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 78, pp. 1–23.
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173.
- Carvalho, C.M.; Gannon, B.W.; Halfhide, D.E.; Santos, S.B.; Hayes, C.M.; Roe, J.M.; Azeredo, J. The in vivo efficacy of two administration routes of a phage cocktail to reduce numbers of Campylobacter coli and Campylobacter jejuni in chickens. BMC Microbiol. 2010, 10, 232.
- Merabishvili, M.; Pirnay, J.-P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-Controlled Small-Scale Production of a Well-Defined Bacteriophage Cocktail for Use in Human Clinical Trials. PLoS ONE 2009, 4, e4944.
- Viazis, S.; Akhtar, M.; Feirtag, J.; Diez-Gonzalez, F. Reduction of Escherichia coli O157:H7 viability on hard surfaces by treatment with a bacteriophage mixture. Int. J. Food Microbiol. 2011, 145, 37–42.
- Geredew Kifelew, L.; Mitchell, J.G.; Speck, P. Mini-review: Efficacy of lytic bacteriophages on multispecies biofilms. Biofouling 2019, 35, 472–481.
- Tian, F.; Li, J.; Nazir, A.; Tong, Y. Bacteriophage—A Promising Alternative Measure for Bacterial Biofilm Control. Infect. Drug Resist. 2021, 14, 205–217.
- Gutiérrez, D.; Rodríguez-Rubio, L.; Martínez, B.; Rodríguez, A.; García, P. Bacteriophages as Weapons Against Bacterial Biofilms in the Food Industry. Front. Microbiol. 2016, 7, 825.
- Van Nieuwenhuyse, B.; Galant, C.; Brichard, B.; Docquier, P.-L.; Djebara, S.; Pirnay, J.-P.; Van der Linden, D.; Merabishvili, M.; Chatzis, O. A Case of In Situ Phage Therapy against Staphylococcus aureus in a Bone Allograft Polymicrobial Biofilm Infection: Outcomes and Phage-Antibiotic Interactions. Viruses 2021, 13, 1898.
- Sanmukh, S.G.; Santos, N.J.; Barquilha, C.N.; dos Santos, S.A.; Duran, B.O.; Delella, F.K.; Moroz, A.; Justulin, L.A.; Carvalho, H.F.; Felisbino, S.L. Exposure to Bacteriophages T4 and M13 Increases Integrin Gene Expression and Impairs Migration of Human PC-3 Prostate Cancer Cells. Antibiotics 2021, 10, 1202.
- Vellon, L.; Menendez, J.A.; Liu, H.; Lupu, R. Up-regulation of αVβ3 integrin expression is a novel molecular response to chemotherapy-induced cell damage in a heregulin-dependent manner. Differentiation 2007, 75, 819–830.
- Bichet, M.C.; Chin, W.H.; Richards, W.; Lin, Y.-W.; Avellaneda-Franco, L.; Hernandez, C.A.; Oddo, A.; Chernyavskiy, O.; Hilsenstein, V.; Neild, A.; et al. Bacteriophage uptake by mammalian cell layers represents a potential sink that may impact phage therapy. iScience 2021, 24, 102287.
- Hsiao, I.-L.; Gramatke, A.M.; Joksimovic, R.; Sokolowski, M.; Gradzielski, M.; Haase, A. Size and cell type dependent uptake of silica nanoparticles. J. Nanomed. Nanotechnol. 2014, 5, 1.
- Ackermann, H.-W.; Tremblay, D.; Moineau, S. Long-term bacteriophage preservation. WFCC Newsl. 2004, 38, 35–40.
More