The inflammasome is an intracellular molecular complex, which is mainly involved in innate immunity. Inflammasomes are formed in response to danger signals, associated with infection and injury, and mainly regulate the secretion of interleukin-1β and interleukin-18. Inflammasome dysregulation is known to be associated with various diseases and conditions, and its regulatory mechanisms have become of great interest in recent years. In the colon, inflammasomes have been reported to be associated with autophagy and the microbiota, and their dysregulation contributes to colitis and. However, the detailed role of inflammasomes in inflammatory bowel disease is still under debate because the mechanisms that regulate the inflammasome are complex and the inflammasome components and cytokines show seemingly contradictory multiple effects.
- NLRP3
- inflammasome
- IL-1β
- IL-18
Note: All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
The innate immune system is activated when a pattern recognition receptor (PRR) sensor recognizes a danger signal. Danger signals that are common to pathogens are referred to as pathogen-associated molecular patterns (PAMPs). In recent years, it has become clear that some PRRs also recognize danger-associated molecular patterns (DAMPs), which are secreted by stressed and injured cells and cause an aseptic inflammatory reaction. Therefore, the pattern recognition mechanism is involved not only in acute inflammation, which is associated with pathogen infections, but also in inflammation associated with various chronic inflammatory diseases and bacteria inside and outside the body. The inflammasome is an intracellular molecular complex, which is mainly involved in innate immunity and is formed in response to danger signals associated with infection and injury. Inflammasome dysregulation is known to be associated with various diseases and conditions, and its regulatory mechanism has become of great interest in recent years. The concept of the inflammasome was proposed in 2002 based on the analysis of cryopyrin-associated periodic syndrome (CAPS), a hereditary autoinflammatory disorder [1]. In 2006, it was reported that inflammation in gout attacks was mediated by inflammasome activation by urate crystals, which became a focus of attention as a new molecular mechanism of aseptic inflammation [2].
PRRs aggregate with apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD) (ASC) and caspase-1 to form the inflammasome, which induces caspase-1 activation. The activated caspase-1 induces cell death, called pyroptosis, and cleaves precursors of inflammatory cytokines, such as interleukin (IL)-1β and IL-18, to induce the extracellular release of their mature forms [3][4][5][6][3–6]. Cytokine secretion is known to be associated with cases of pyroptosis due to caspase-1 activation or extracellular release of organelles. Caspase-1 cleaves gasdermin D to form cell membrane pores, causing pyroptosis, which is accompanied by the outflow of cytoplasmic components and induces an inflammatory response by releasing mature inflammatory cytokines [7]. Thus, inflammasomes play a role in defense against infection by inducing both immune activation by inflammatory cytokines and the elimination of infected cells via pyroptosis.
Several types of inflammasomes have been reported so far, among which the Nod-like receptor (NLR) family pyrin domain (PYD)-containing 3 (NLRP3) inflammasome is activated not only by PAMPs but also by DAMPs and can cause aseptic inflammation in various diseases, such as gout and arteriosclerosis. In the intestine, inflammasomes are not only important mediators of the host defense but also important regulators of intestinal homeostasis, which regulate the defense function of the intestinal epithelium and the immune response to the gut microbiota [8][9][10][8–10]. Dysregulation of the inflammasome is known to be associated with various diseases. In the intestine, inflammasomes are known to be associated with autophagy and the microbiota, and dysregulation of inflammasomes contributes to the development of inflammatory bowel disease (IBD). Among all kinds inflammasome, the NLRP3 inflammasome is the most characterized one. The NLRP3 inflammasome, has been analyzed extensively in its contribution to colitis and is considered to be important in the development of therapeutic strategies. However, the detailed role of inflammasomes in IBD is still under debate because the mechanism that regulates the inflammasome is complex and the inflammasome components and cytokines show seemingly contradictory multiple effects. In addition, the contribution of NLRP3 inflammasome to intestinal inflammation is complicated, because recent experimental data suggest that the manipulation of microbiota in NLRP3 knockout (KO) mice cause contradictory data.
2. Structure of the Inflammasome
The basic structure of the inflammasome consists of a PRR as a sensor molecule, ASC as an adapter molecule, and caspase-1 [11]. PRRs include Toll-like receptors (TLRs), NLRs, the retinoic acid-inducible gene-I-like receptor (RLR), and the pyrin and HIN domain (PYHIN) receptor family which includes the absent in melanoma 2 (AIM2) -like receptor (ALR) and C-type lectins. In the NLR subfamily, NLRP1, NLRP3, NLR family CARD-containing 4 (NLRC4), NLRP6, and NLRP12 are commonly known [12][13][12,13].
Each of the constituent molecules of the inflammasome has a PYD and CARD, which are domain structures responsible for the characteristic intermolecular interactions and interact in a homophilic binding mode. For example, the structure of the NLRP3 inflammasome is presented in Figure 1. In the NLRP3 protein, there are three core domains [1][2][1,2]. The NACHT [NAIP (neuronal apoptosis inhibitory protein), CIITA (MHC class II transcription activator), HET-E (incompatibility locus protein from Podospora anserina), and TP1 (telomerase-associated protein)] domain promotes self-oligomerization and ATPase activity. NACHT is required for the activation of inactive procaspase-1 into active caspase-1 and subsequent homo- or heterooligomerization, leading to autocleavage and secretion of IL-1β and IL-18. The N-terminus contains a PYD, which binds to caspase-1 via an adapter protein (ASC) to form a complex. The C-terminus contains a leucine-rich repeat (LRR), which plays an essential role in the responsiveness to danger signals. Inflammasome assembly is inhibited by the LRR domain, whose activity is neutralized by the activation signal from PAMPs or DAMPs. ASC includes two transduction domains, a PYD, which can connect to the upstream NLRP3, and a CARD, which can connect to the downstream caspase-1.
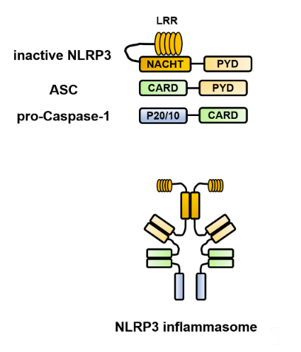
Figure 1. Structure of the Nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome. NLRP3 self-polymerizes via the NACHT [NAIP (neuronal apoptosis inhibitory protein), CIITA (MHC class II transcription activator), HET-E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase-associated protein)] domain and binds to apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) via the pyrin domain (PYD). Furthermore, ASC binds to caspase-1 via the caspase recruitment domain (CARD) to form a complex, which brings the caspase-1 precursors into close proximity to each other, leading to their self-activation and processing of pro-interleukin (IL)-1β and pro-IL-18 into their mature forms. ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; CARD, caspase recruitment domain; IL, interleukin; LRR, leucine-rich repeat; NLRP3, Nod-like receptor family pyrin domain-containing 3; NACHT, (neuronal apoptosis inhibitory protein, MHC class II transcription activator, incompatibility locus protein from Podospora anserina, and telomerase-associated protein); PYD, pyrin domain.
3. Control Mechanism of the NLRP3 Inflammasome Activation
3.1. Two-Signal Control of the NLRP3 Inflammasome Activation
The activation of the NLRP3 inflammasome implicates a first signal, called priming, which requires an inflammatory stimulus involved in transcriptional induction, and a second signal, called triggering, which requires a danger signal involved in posttranslational regulation [8][11][14][8,11,14] (Figure 2). These mechanisms are distinctive for the IL-1 family and are different from those involved in the production of other cytokines.
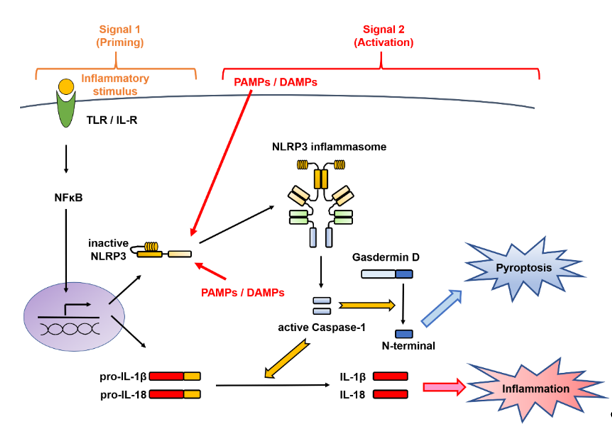
Figure 2. Two-signal control of the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation. Activation of the NLRP3 inflammasome implicates a first signal, called priming, which requires an inflammatory stimulus involved in transcriptional induction, and a second signal, called triggering, which requires a danger signal involved in posttranslational regulation. In the first stage is priming signal for inflammasome activation. mRNA expression of the interleukin (IL-1β)- and NLRP3-encoding genes from Toll-like receptor (TLR) or the IL-1 receptor (IL-1R) through nuclear factor-kappa B (NF-κB) and NLRP3 protein expression by deubiquitination are induced. In the second stage, microbial or danger signals can directly activate inflammasome assembly. DAMPs, danger-associated molecular patterns; IL, interleukin; IL-1R, interleukin-1 receptor; NF-κB, nuclear factor-kappa B; NLRP3, Nod-like receptor family pyrin domain-containing 3; PAMPs, pathogen-associated molecular patterns; TLR, Toll-like receptor.
3.2. NLRP3 Inflammasome Regulators
Each PRR recognizes a different danger signal [15][16][15,16] (Figure 3). Since pathogen-derived PAMPs mainly activate these PRRs, the latter are considered to function as a defense against infection. On the other hand, NLRP3 is activated not only by PAMPs but also by endogenous or exogenous DAMPs and, therefore, is considered of importance in aseptic inflammation. Since there are various stimulators of NLRP3, it is considered that NLRP3 is activated by recognizing a common signal induced by danger signals.
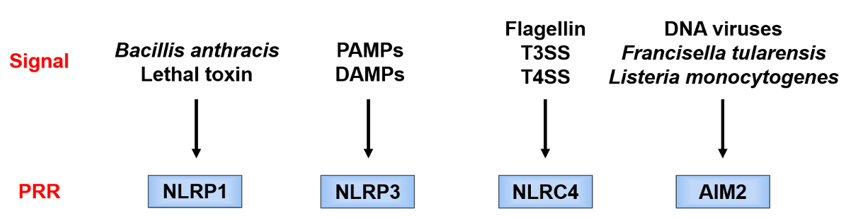
Figure 3. Danger signals recognized by different pattern recognition receptor (PRRs). Each PRR recognizes a different danger signal. NOD-like receptor family pyrin domain-containing 1 (NLRP1) is activated by certain bacterial toxins. NOD-like receptor family caspase recruitment domain-containing 4 (NLRC4) works in combination with neuronal apoptosis inhibitory protein (NAIP), and the ligand itself binds to NAIP. NAIP recognizes the bacterial protein flagellin and the constituent proteins of the type III secretion system (T3SS) of Gram-negative bacteria and forms a complex with NLRC4. Absent in melanoma 2 (AIM2) directly recognizes double-stranded DNA and is considered to be activated during infection with double-stranded DNA viruses, such as cytomegalovirus, and cell-invasive bacteria, such as Francisella tularensis and Listeria monocytogenes. AIM2, absent in melanoma 2; DAMPs, danger-associated molecular patterns; T3SS/T4SS, type III/IV secretion system; PAMPs, pathogen-associated molecular patterns; PRR, pattern recognition receptor; NLRC4, Nod-like receptor family caspase recruitment domain-containing 4; NLRP1/NLRP3, Nod-like receptor family pyrin domain-containing 1/3; NAIP, neuronal apoptosis inhibitory protein.
NLRP3 inflammasome activation involves various types of PAMPs or DAMPs [4][17][4,17]. In addition, the major mechanistic pathways and stimuli that trigger NLRP3 inflammasome activation have been reported to include intracellular K+ efflux, mitochondrial reactive oxygen species (ROS), and lysosomal damage [14] (Figure 4).
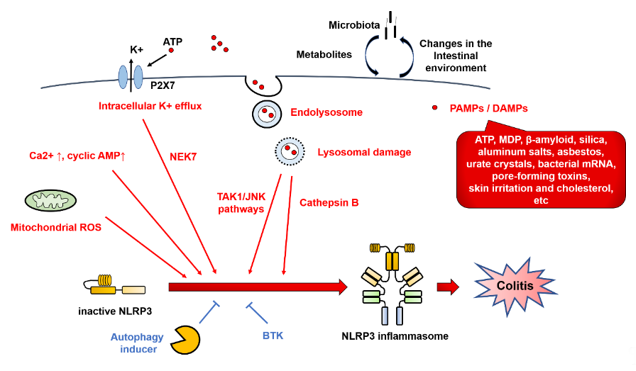
Figure 4. NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome regulators. NLRP3 inflammasome activation involves various types of pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs), such as ATP, muramyl dipeptide (MDP), β-amyloid, silica, asbestos, urate crystals, bacterial mRNA, pore-forming toxins, skin irritants, and cholesterol. In addition, the major mechanistic pathways and stimuli that trigger NLRP3 inflammasome activation have been reported to include intracellular K+ efflux, mitochondrial reactive oxygen species (ROS), and lysosomal damage. Red arrows indicate activation of NLRP3 inflammasome. Blue T-shaped lines indicate inhibition of NLRP3 inflammasome activation. BTK, Bruton’s tyrosine kinase; DAMPs, danger-associated molecular patterns; JNK, c-Jun N-terminal kinase; MDP, muramyl dipeptide; NEK7, NIMA-related kinase 7; NLRP3, Nod-like receptor family pyrin domain-containing 3; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; TAK1, TGF-β-activated kinase 1.
In the intracellular K+ efflux pathway, extracellular ATP, which is released from dead cells, causes K+ efflux via the P2 × 7 receptor, and selective K+ efflux is also caused by bacterial toxins, resulting in a decrease in the intracellular K⁺ concentration and activation of NLRP3. By contrast, NLRP3 activation is blocked by inhibiting K+ efflux from cells [18]. Meanwhile, the opening of large mitochondrial pores is not required for NLRP3 activation [19].
Lysosomal damage is triggered by endogenous uric acid crystals and cholesterol crystals or by exogenous particles, such as silica and asbestos. NLRP3 is activated by the intracellular release of cathepsin B, which is a lysosomal enzyme. The TGF-β-activated kinase 1 (TAK1)/c-Jun N-terminal kinase (JNK) pathway, which is a mitogen-activated protein kinase signaling pathway, is activated via lysosomal disruption, and this kind of activation is required for full activation of the NLRP3 inflammasome [20]. Silica crystals and aluminum salts activate NLRP3 inflammasomes via destabilization of phagosomes. Acidification of phagosomes or inhibition of cathepsin B activity impairs NLRP3 activation [21].
ROS derived from organelles positively regulate NLRP3 inflammasome activity. Mitochondria are a major intracellular source of ROS, and thus, mitochondrial function plays an important role in modulating inflammation. Mitochondrial ROS are produced when inhibition of the electron transport chain or removal of abnormal mitochondria via autophagy (mitophagy) is impaired [22]. Mitochondrial ROS are required for non-transcriptional priming of NLRP3, and NLRP3 deubiquitination is a prerequisite for activation [23]. On the other hand, it has been reported that ROS inhibitors block priming of the NLRP3 inflammasome but not its activation [24].
In addition, NIMA-related kinase 7 (NEK7), protein kinase R, and guanylate-binding protein 5 (GBP5) link these common pathways with NLRP3 [11][25][11,25]. NEK7 is an essential protein that acts downstream of the K+ efflux pathway to mediate NLRP3 inflammasome assembly and activation [26]. NEK7 is involved in the regulation of the cell cycle, mitotic spindle formation, and cytokinesis. Activation of the NLRP3 inflammasome by NEK7 is restricted to the interphase of the cell cycle [27].
Other regulatory mechanisms have been reported to involve NLRP3 phosphorylation [28] and ubiquitination [29], as well as the calcium-sensing receptor (CaSR). The CaSR modulates NLRP3 inflammasome activation via changes in intracellular Ca2+ and cyclic AMP concentrations [30]. Activation of proteasome- and lysosome-dependent mechanisms by the CaSR promotes the degradation of key modulators of NLRP inflammasome activation [31]. Recently, Bruton’s tyrosine kinase (BTK) has been reported to play a role in physiological inhibition of NLRP3 inflammasome activation, which explains why patients with X-linked agammaglobulinemia tend to develop Crohn’s disease (CD) [32]. The platelet-activating factor receptor regulates colitis-induced lung inflammation via the NLRP3 inflammasome [33]. Kynurenic acid/GPR35 axis limits NLRP3 inflammasome activation and exacerbates colitis in socially stressed mice [34]. These findings suggest that NLRP3 inflammasome activation involves a wide range of regulatory mechanisms.
3.3. Non-Canonical Inflammasomes
In recent years, new protein complexes, called non-canonical inflammasomes, have ben discovered. Caspase-11 plays a central role in mice, while caspase-4 and caspase-5 play a central role in humans. Caspase-11 is critical for caspase-1 activation and IL-1β production in macrophages infected with Escherichia coli and Citrobacter rodentium [35][36][35,36]. Lipopolysaccharide (LPS) or outer membrane vesicle derived from Gram-negative bacteria enter host cells via several different routes, leading to the direct interaction with caspase-11 to form LPS-caspase-11 complexes. This promotes inflammasome-independent pyroptosis and proteolytic activation of caspase-1 and activation of caspase-1 to cleave pro-IL-1β and pro-IL-18 into secreted bioactive cytokines [37].
References
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426, doi:10.1016/s1097-2765(02)00599-3.
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241, doi:10.1038/nature04516.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832, doi:10.1016/j.cell.2010.01.040.
- Aguilera, M.; Darby, T.; Melgar, S. The complex role of inflammasomes in the pathogenesis of Inflammatory Bowel Diseases—Lessons learned from experimental models. Cytokine Growth Factor Rev. 2014, 25, 715–730, doi:10.1016/j.cytogfr.2014.04.003.
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Rev. Immunol. 2009, 27, 519–550, doi:10.1146/annurev.immunol.021908.132612.
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Rev. Immunol. 2013, 13, 397–411, doi:10.1038/nri3452.
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Rev. 2017, 277, 61–75, doi:10.1111/imr.12534.
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Pharmacol. 2015, 6, 262, doi:10.3389/fphar.2015.00262.
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Immunol. 2019, 10, 276, doi:10.3389/fimmu.2019.00276.
- Shao, B.Z.; Wang, S.L.; Pan, P.; Yao, J.; Wu, K.; Li, Z.S.; Bai, Y.; Linghu, E.Q. Targeting NLRP3 Inflammasome in Inflammatory Bowel Disease: Putting out the Fire of Inflammation. Inflammation 2019, 42, 1147–1159, doi:10.1007/s10753-019-01008-y.
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Biochem. Sci. 2016, 41, 1012–1021, doi:10.1016/j.tibs.2016.09.002.
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Rev. Immunol. 2016, 16, 407–420, doi:10.1038/nri.2016.58.
- Ranson, N.; Kunde, D.; Eri, R. Regulation and Sensing of Inflammasomes and Their Impact on Intestinal Health. J. Mol. Sci. 2017, 18, 2379, doi:10.3390/ijms18112379.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. J. Mol. Sci. 2019, 20, 3328, doi:10.3390/ijms20133328.
- Place, D.E.; Kanneganti, T.D. Recent advances in inflammasome biology. Opin. Immunol. 2018, 50, 32–38, doi:10.1016/j.coi.2017.10.011.
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. Cell Biol. 2016, 213, 617–629, doi:10.1083/jcb.201602089.
- Opipari, A.; Franchi, L. Role of inflammasomes in intestinal inflammation and Crohn's disease. Bowel. Dis. 2015, 21, 173–181, doi:10.1097/MIB.0000000000000230.
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589, doi:10.1038/sj.cdd.4402195.
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153, doi:10.1016/j.immuni.2013.05.016.
- Okada, M.; Matsuzawa, A.; Yoshimura, A.; Ichijo, H. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. Biol. Chem. 2014, 289, 32926–32936, doi:10.1074/jbc.M114.579961.
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Immunol. 2008, 9, 847–856, doi:10.1038/ni.1631.
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910, doi:10.1016/j.cell.2015.12.057.
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. Biol. Chem. 2012, 287, 36617–36622, doi:10.1074/jbc.M112.407130.
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Núñez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. Immunol. 2011, 187, 613–617, doi:10.4049/jimmunol.1100613.
- Shenoy, A.R.; Wellington, D.A.; Kumar, P.; Kassa, H.; Booth, C.J.; Cresswell, P.; MacMicking, J.D. GBP5 promotes NLRP3 inflammasome assembly and immunity in mammals. Science 2012, 336, 481–485, doi:10.1126/science.1217141.
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357, doi:10.1038/nature16959.
- Shi H.; Wang Y.; Li X.; Zhan X.; Tang M.; Fina M.; Su L.; Pratt D.; Bu C.H.; Hildebrand S.; et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Immunol. 2016, 17, 250–258, doi: 10.1038/ni.3333.
- Gong T.; Jiang W.; Zhou R. Control of Inflammasome Activation by Phosphorylation. Biochem. Sci. 2018, 43, 685–699, doi: 10.1016/j.tibs.2018.06.008.
- Py B.F.; Kim M.S.; Vakifahmetoglu-Norberg H.; Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Cell 2013, 49, 331–338, doi: 10.1016/j.molcel.2012.11.009.
- Lee G.S.; Subramanian N.; Kim A.I.; Aksentijevich I.; Goldbach-Mansky R.; Sacks D.B.; Germain R.N.; Kastner D.L.; Chae J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127, doi: 10.1038/nature11588.
- Gutiérrez-López T.Y.; Orduña-Castillo L.B.; Hernández-Vásquez M.N.; Vázquez-Prado J.; Reyes-Cruz G. Calcium sensing receptor activates the NLRP3 inflammasome via a chaperone-assisted degradative pathway involving Hsp70 and LC3-II. Biochem. Biophys. Res. Commun. 2018, 505, 1121–1127, doi: 10.1016/j.bbrc.2018.10.028.
- Mao L.; Kitani A.; Hiejima E.; Montgomery-Recht K.; Zhou W.; Fuss I.; Wiestner A.; Strober W. Bruton tyrosine kinase deficiency augments NLRP3 inflammasome activation and causes IL-1β-mediated colitis. Clin. Investig. 2020, 130, 1793–1807, doi: 10.1172/JCI128322.
- Liu G.; Mateer S.W.; Hsu A.; Goggins B.J.; Tay H.; Mathe A.; Fan K.; Neal R.; Bruce J.; Burns G.; et al. Platelet activating factor receptor regulates colitis-induced pulmonary inflammation through the NLRP3 inflammasome. Immunol. 2019, 12, 862–873, doi: 10.1038/s41385-019-0163-3.
- Zheng X.; Hu M.; Zang X.; Fan Q.; Liu Y.; Che Y.; Guan X.; Hou Y.; Wang G.; Hao H. Kynurenic acid/GPR35 axis restricts NLRP3 inflammasome activation and exacerbates colitis in mice with social stress. Brain Behav. Immun. 2019, 79, 244–255, doi: 10.1016/j.bbi.2019.02.009.
- Kayagaki N.; Warming S.; Lamkanfi M.; Vande Walle L.; Louie S.; Dong J.; Newton K.; Qu Y.; Liu J.; Heldens S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121, doi: 10.1038/nature10558.
- Rathinam V.A.; Vanaja S.K.; Waggoner L.; Sokolovska A.; Becker C.; Stuart L.M.; Leong J.M.; Fitzgerald K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619, doi: 10.1016/j.cell.2012.07.007.
- Yi Y.S. Caspase-11 Non-Canonical Inflammasome: Emerging Activator and Regulator of Infection-Mediated Inflammatory Responses. J. Mol. Sci. 2020, 21, 2736, doi: 10.3390/ijms21082736.