2. Clinical Presentation
It is essential to record the clinical findings and their course as accurately as possible. Therefore, a thorough exploration of the patient’s history of symptoms and a complete physical and neurological examination should be considered as the first step in the diagnosis of ALS.
The clinical hallmarks of ALS are related to the impairment of voluntary muscles, resulting in progressive weakness of the limbs, speech and swallowing dysfunction and respiratory failure with concomitant signs, such as muscle atrophy, fasciculations and increased muscle tone. These motor dysfunctions are derived from the combined impairment of the upper motor neurons (UMN) in the motor cortex of the brain and the lower motor neurons (LMN) in the brainstem and the spinal cord
[1][3][1,3]. Though generally unaffected, there is also evidence of oculomotor, sphincter and autonomic dysfunction
[4][5][6,7]. Initially, the disease typically presents itself either with asymmetrical focal muscle weakness in the upper or lower limbs in spinal onset ALS or with speech and swallowing difficulties due to facial, tongue and pharyngeal muscle weakness in bulbar-onset ALS
[2]. Disease propagation preferably follows an organized contiguous pattern with first symptoms in one limb, subsequently spreading to the contralateral limb and, later, to adjacent regions
[6][8]. However, disease progression is highly variable and frequently shows a non-linear decline
[7][9]. The anatomically separated body regions (or segments) are defined as the bulbar, cervical, thoracic and lumbar regions, with the latter three comprising the spinal regions
[8][10].
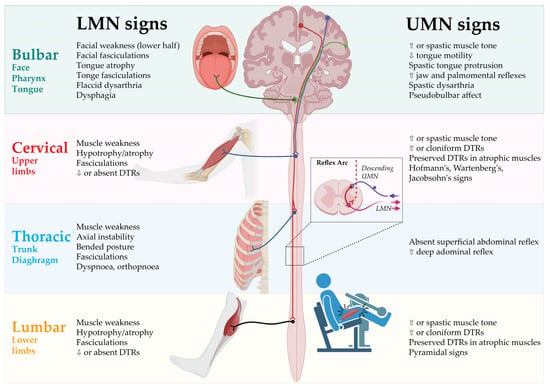
The clinical signs of UMN and LMN involvement according to each of the body regions are presented in
Figure 1.
Figure 1. Clinical signs of UMN and LMN involvement according to body regions. DTR: Deep tendon reflex; LMN: Lower motor neurons; UMN: Upper motor neurons. Created by Castro-Gomez with “
BioRender.com”; accessed on 4 February 2023.
The notable clinical heterogeneity of motor manifestations frequently leads to controversy in the description of phenotypes as opposed to the determination of the diagnosis of ALS. Distinguishing these phenotypes is of relevance, as they are associated with various disease progression rates and survival times
[9][11].
The different clinical phenotypes of ALS are generally described with regard to the extent of UMN and LMN impairment, its distribution and progression to the different body segments
[2][9][2,11].
2.1. Spinal-Onset ALS
ALS with spinal onset is defined by focal weakness in distal muscle groups of the limbs and simultaneous UMN and LMN involvement. With up to 82% of all ALS patients
[10][12], it is the most common phenotype, termed typical or classical ALS. Distal segments of the upper or lower limbs are affected in a focal manner at the onset of the disease. Characteristically, thenar eminence with the abductor pollicis brevis (APB) and the first dorsal interosseus (FDI) muscles are more affected compared to the hypothenar muscle abductor digiti minimi (ADM), referred to as split-hand syndrome
[11][13]. Onset in the dominant hand is thought to be predominant
[12][14]. Lower-limb weakness typically becomes apparent as it causes an unsteady gait due to weak foot dorsiflexion
[13][15]. Notably, UMN dysfunction is not always easily identified in wasted or atrophic muscles of the limbs, particularly in the early stages of the disease
[14][15][16,17].
2.2. Bulbar-Onset ALS
ALS with bulbar onset, or bulbar ALS, is the second leading phenotype with initial motor dysfunction in the bulbar region. Speech difficulties and frequent choking with concomitant hypersalivation are the cardinal presenting symptoms. Both LMN and UMN impairment are present, causing tongue wasting with fasciculations, facial spasticity and pseudobulbar affect in the early stages of the disease. Propagation to other spinal regions is evident later in the disease’s course
[16][18]. Bulbar UMN involvement, also known as pseudobulbar palsy, can become clinically indicated by emotional lability, accompanied by excessive crying or laughing response to minor stimuli. This is termed the “pseudobulbar affect”. Other bulbar UMN signs include facial spasticity and slowed spastic movement of the tongue, whereas slurred speech, wasting of the tongue and fasciculations indicate bulbar LMN impairment
[1][3][1,3]. Frontal release signs, such as the palmomental reflex, may also indicate bulbar involvement
[17][19]. In contrast to the often used phrase “progressive bulbar palsy”, “ALS with bulbar onset” appears to be the more convenient term, as it represents a phenotypic description rather than a diagnostic label, suggesting no progression to other body regions
[9][11].
2.3. Progressive Muscular Atrophy
Progressive muscular atrophy (PMA) presents with clinically isolated LMN impairment of the anterior horn cells and brainstem motor nuclei. However, subclinical UMN impairment can be detected in the early stages of the disease
[18][20]. Although PMA tends to have a slower disease progression than classical ALS
[19][21], 20 to 30% of the patients may develop ALS with clinically evident UMN impairment within 5 to 10 years from the disease’s onset. It is still controversially debated whether PMA should be considered a variant of ALS
[18][20].
2.4. Primary Lateral Sclerosis
Primary lateral sclerosis (PLS) is characterized by progressive isolated UMN dysfunction detectable in at least two regions (thoracic region will not be considered) for at least two years. LMN dysfunction is absent, whereas minimal signs of denervation (positive sharp waves or fibrillation potentials) on electromyography (EMG) are permitted. Whether PLS constitutes a separate disease entity or rather represents a clinically benign variant of ALS remains controversial
[20][22].
2.5. Flail-Arm-Syndrome
In flail-arm-syndrome (FAS), also known as Vulpian Bernhardt’s type, progressive, proximal and symmetrical weakness of both upper limbs caused by LMN impairment is predominantly apparent. Motor symptoms in bulbar muscles or lower limbs are unaffected from 12 to 20 months after the onset of upper limb symptoms
[21][22][23,24]. LMN involvement is predominant, whereas UMN involvement can be occasionally present in lower limbs
[23][25]. FAS represents a rather benign phenotype with a median survival time of 4 years and a 10 year survival rate of 17%
[21][23].
2.6. Flail-Leg-Syndrome
Analogous to FAS, flail-leg-syndrome (FLS) presents itself with progressive and symmetrical weakness of both lower limbs, whereas distal muscle groups are typically affected and LMN involvement outweighs UMN involvement
[22][24]. Other segments are clinically spared for a mean of 16 months after the disease’s onset. Unlike FAS, FLS has a similar prognosis as spinal-onset ALS, with a median survival time of 3 years and a 10-year survival rate of 13%
[21][23].
2.7. Axial or Respiratory-Onset ALS
Axial-onset ALS initially presents itself with weakness of trunk muscles. Typically, paravertebral muscles are affected, resulting in bent posture, axial instability and dropped head syndrome
[24][26]. However, weakness of the thoracic muscles can be rather difficult to recognize
[6][8].
In respiratory-onset ALS patients suffer from dyspnoea and orthopnoea at the beginning of the disease, caused by weakness of the respiratory muscles and the diaphragm, which is also anatomically related to the thoracic region
[25][27]. The prognosis is poor due to early respiratory failure and complications such as pneumonia
[21][24][26][23,26,28].
There is no reliable method to detect thoracic UMN involvement
[27][29]. However, brisk and deep abdominal reflexes, particularly with diminished or absent superficial abdominal reflexes, might be suggestive signs
[28][30].
2.8. Hemiplegic ALS (Mill’s Syndrome)
This very rare phenotype is defined by slowly progressive, unilateral muscle weakness in the limbs alongside clinically predominant UMN signs, such as pathological deep tendon reflexes (DTR) and pyramidal tract signs. The onset may either occur in the upper limbs with subsequent descending propagation to the lower limbs or vice versa
[29][31]. As is the case with PLS, there is an ongoing debate regarding whether Mill’s syndrome should be considered a distinct clinical entity in the spectrum of motor neuron diseases or an ALS variant
[29][30][31][31,32,33].