The International Headache Society (IHS) issued an International Headache Classification (ICHD)HD III report, the third edition of the International Headache Classification, which defines alcohol-induced headaches (AIHs) as headaches that occur immediately after or a while longer after ingesting alcoholic beverages. The ICHD report classifies AIHs into two categories. The classification parameter is the time lapse between consumption and headache manifestation: (1) AIH manifestation within 3 h after alcohol consumption is termed immediate alcohol-induced headache, and (2) manifestations after 5–12 h are termed delayed alcohol-induced headache. Whether an AIH effect manifests immediately or is delayed depends on groups, regions, and the specific alcoholic beverages consumed. Alcohol-induced headaches (AIHs) as an important underlying factor for neuroinflammation. Studies show the relationship between alcoholic beverages, AIH agents, neuroinflammation, and the pathway they elicit.
1. Ethanol and Its Metabolism In Vivo
1.1. Transient Receptor Potential Cation Channel Subfamily V Member RPV1
Transient receptor potential cation channel, subfamily V, member 1 (TRPV1), also known as the capsaicin/heat receptor and the vanilloid receptor, is a confirmed pain trigger mentioned in numerous studies. Several inflammatory studies have shown that the expression of TRPV1 is upregulated in
alcohol-induced headaches (AIH
) animal models
[1][2][29,30]. Meanwhile, several studies have reported phenomena such as greater alcohol consumption and weaker withdrawal severity in TRPV1
−/− mutant mice
[3][4][5][6][7][31,32,33,34,35]. These experiments implicate TRPV1 for the alcoholic beverages’ consumption hangover symptoms and AIH effects.
Nicoletti et al.
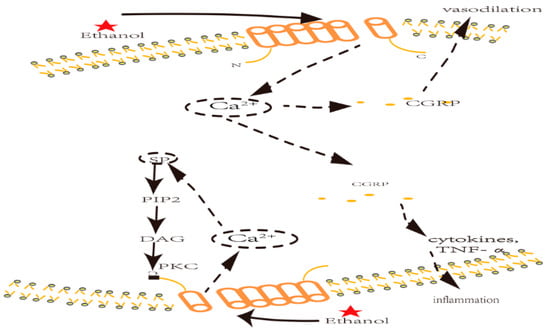
[8][9] demonstrated that ethanol-induced neurogenic inflammation in the trigeminal system and meningeal vessels vasodilation are caused by the activation of TRPV1 and the release of calcitonin gene-related peptide (CGRP)
[9][36] and substance P (SP)
[10][37]. They measured CGRP and SP in a group treated with a TRPV1 antagonist and found that CGRP and SP were decreased by 70% compared with the control group under the same ethanol treatment.
CGRP has been acknowledged
[11][38] as a trigger for vasodilation through mitogen-activated protein kinase (MAPK) activation, as well as an inflammatory mediator for cytokines (i.e., TNF-α
[12][39]). An in vivo study indicates that CGRP triggers vasodilation in porcine coronary arteries
[10][37]. Blocking TRPV1 with an antagonist—capsazepine—resulted in lower CGRP expression and decreased arterial dilatation. It could be explained that TRPV1 sensitization first elicits a Ca
2+ influx, and Ca
2+ then triggers a rapid downstream signaling pathway resulting in the release of inflammatory mediators such as CGRP and SP (see
Figure 1). The release of these neuropeptides can be suppressed if Ca
2+ is removed
[8][9], supporting the notion that ethanol promotes Ca
2+ inflow. Inflammatory mediators, such as CGRP and SP, promote the downstream cascade of neuroinflammation factors and cytokines, which culminate in neuroinflammation.
SP can also sensitize TRPV1, which is associated with phosphatidylinositol-4,5-bisphosphate (PIP2) cleavage, diacylglycerol (DAG) production, and the activation of protein kinase C (PKC). According to some studies
[13][14][40,41], PKC can activate TRPV1 at its C terminus. Ethanol treatment upregulated PKCε expression in Western blotting and plasma membrane tests. These results reveal that PKC is an important TRPV1 sensitization ligand
[15][42].
Peralta et al. found that TRPV1 mRNA was upregulated in alcohol-treated 5-month-old wild-type mice relative to untreated mice, suggesting that ethanol was significantly associated with TRPV1 expression. The same study showed that the activation of TRPV1 was markedly associated with c-Jun N-terminal kinase (JNK) and p38 MAPK (modulated by miR-183c and miR-200s)
[16][43]. Sakakibara and colleagues compared oral epithelial cells from patients with different levels of alcohol consumption. Patients with a history of lengthier alcohol consumption had significantly higher TRPV1 mRNA expression levels in their epithelial cells than patients without this history
[17][44].
However, changes in TRPV1 protein levels have not been observed in response to ethanol treatment. Ethanol treatment (0.3% concentration) did not affect TRPV1 protein levels in either the plasma membrane or the total cell proteins of HEK293 cells
[18][45]. Additionally, acetate may also cause
alcohol-induced headaches (AIHs
) through the release of adenosine. However, this effect can be blocked by the anti-inflammatory drug ketorolac
[19][46]. Additional evidence suggests that adenosine induces hyperalgesia through TRPV1 phosphorylation. This result implies that acetate may also affect AIHs through TRPV1
[20][21][47,48]. Thus, ethanol and acetate may affect AIHs by activating TRPV1. Additionally, ethanol may affect TRPV1 activity through its sensitization rather than upregulation.
1.2. Toll-Like Receptor R4
Toll-like receptor 4 (TLR4) expression is also associated with AIHs, and its effects are ascribed to neuroinflammation
[22][49]. Evidence suggests that ethanol can affect TLR4-induced neuroinflammation, which then elicits AIHs. A study showed that TLR4 knockout mice exhibited less ethanol-induced inflammatory damage
[23][50]. The result supports the motion that TLR4 has AIH effects.
Unlike TRPV1, TLR4 affects changes in gene expression, protein abundance
[24][25][51,52], and activation
[26][27][53,54] in response to ethanol and its metabolites. These results are confirmed in many studies. It was confirmed that miR-125b (the upstream miRNA for TLR4) was upregulated in alcohol-treated wild-type mice compared with untreated mice. Ethanol elicited upregulation of TLR4 mRNA
[16][43]. A study found that ethanol upregulated TLR4 mRNA and protein expression levels in the microglial cell membrane and total cell protein. Maximal TLR4 mRNA levels were observed after 30 min of ethanol treatment
[24][51].
Ethanol treatment elicits high levels of TNF-α and IL-1β after 7–24 h. High levels of inductive nitric oxide synthase (iNOS) and cyclo-oxygenase (COX-2) are observed 3–7 h after ethanol treatment, accompanied by the upregulation of TLR4
[28][55]. A team observed that treating mice with 6 g/kg ethanol increased mRNA levels of chemokines, such as CCL2, MCP-1, and IL-6
[29][56]. Another study found that interleukin (IL) 6, IL-10, and IL-12 were upregulated via TLR4 after mice models were treated with 6 g/kg ethanol
[30][57].
A study showed that the TRL4 inflammation downstream factors, such as TNF-α and IL-1β, were reduced following ethanol exposure in the TLR4
−/− mutant relative to the wild type. Another research demonstrated that ethanol did not affect TNF-α, IL-1β, COX-2, and iNOS in the cerebral cortex of the TLR4
−/− mutant
[31][58]. Additionally, the levels of NF-κB, COX-2, MyD88, CD40, and TRIF were suppressed, and neuropathic pain was attenuated in animal models when TLR4 was blocked by butylated hydroxytoluene (BHT) and
Rhodobacter sphaeroides’ lipopolysaccharide (LPS-RS)
[17][32][44,59].
Many studies confirm that the upregulation of TLR4 delays AIH manifestation. The release of inflammation factors, e.g., TNF-α, IL-1β, COX-2, and iNOS, were significantly affected by ethanol treatment and TLR4 overexpression. In contrast, the release time of these substances was closely associated with AIHs, with a 5–12 h delay
[33][60].
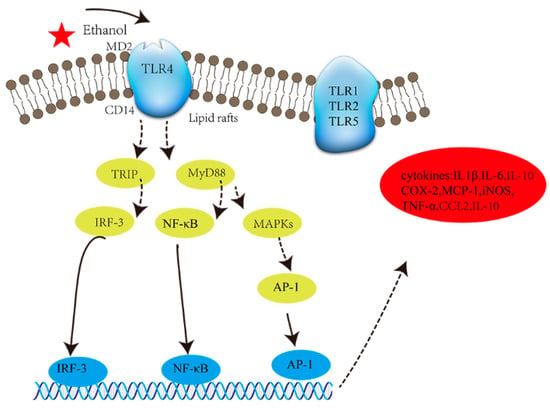
Activated TLR4 co-localizes with adaptor proteins (i.e., CD14 and MD2, see
Figure 1) in lipid rafts
[25][52]. TLR4 transmits signals to adaptor protein molecules such as TIR-domain-containing adaptor-inducing IFNβ (TRIF) and myeloid differentiation primary response gene88 (MyD88). Both of these adaptor proteins induce the activation of transcription nuclear factors (TnFs) such as NF-κB, activator protein 1 (AP-1), and interferon regulatory factor 3 (IRF3). Eventually, TnFs promote the release of cytokines such as IL-1β
[34][35][61,62], TNF-α
[36][63], MCP-1, and inflammatory mediators such as COX-2 and iNOS
[28][55]. The expression of the aforementioned induces inflammation (
Table 1).
Figure 1. The activation of TLR4. Key: MD2: myeloid differential protein 2; CD14: membrane CD14; MAPK: mitogen-activated protein kinase; MCP-1: monocyte chemoattractant protein-1; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α; IL1β: interleukin-1β; CCL2: chemokine ligand 2.
Ethyl-b-D-glucuronide (EtG) is a minor metabolite of ethanol. EtG makes up 0.04% of ethanol derivatives
[37][64] and has a similar structure to glucuronic acid (GA)
[38][65]. It elicits pain through TLR4 sensitization. Lewis et al.
[39][66] studied the effects of ethanol and EtG on TLR4. These compounds sensitize TLR4 and elicit allodynia (a rat model of AIHs) when administered by intrathecal injection. They also induce the overexpression of secreted alkaline phosphatase (SEAP) in Sprague Dawley rats. TLR4 antagonists reduced this allodynia and SEAP secretion by blocking TLR4.
Table 1.
Research on the effects of ethanol and its derivatives on AIHs and neuroinflammation in vivo.
2. Effects of Ethanol and Its Metabolites In Vitro
2.1. Transient Receptor Potential Cation Channel Subfamily V Member RPV1
A previous study suggested that ethanol could increase capsaicin sensitivity by decreasing the thermal threshold of the TRPV1 channel in human embryonic kidney 293 (HEK 293) cells from −42 to −34 °C. Advances in the research field have allowed the ethanol effect to be studied on the expression of certain proteins. A study elucidated the effects of ethanol on the expression of the transient receptor potential (TRP) channel in brain microvascular endothelial cells (BMVECs). Although they did not find expression of TPRV1 in BMVECs, ethanol upregulated TRPV2 and TRPV4 mRNA and protein levels
[43][70], which increased 2 h after treatment with 17.4 mm ethanol. A related work demonstrated that TRPV1 mRNA was upregulated by ethanol treatment
[16][17][43,44]. However, no significant changes in TRPV1 protein levels were observed between control and ethanol-treated cells
[18][45] (
Table 2).
TRPV1 can be potentiated by several triggers, including protein kinase A (PKA)
[44][71] and PKC
[45][72]. Most in vivo studies indicate that ethanol directly activates TRPV1 via PKC
[10][46][37,73]. However, TRPV1 was activated through PIP2 versus PKC in HEK293 cells
[18][45], and the same study showed that depletion of PIP2 limited TRPV1 sensitization by ethanol. Although no unified theory has emerged to describe the relationship between TRPV1 sensitization and the ethanol-inducible pathway, in vitro and in vivo experiments mentioned above suggest that ethanol may activate TRPV1 through the Ca
2+-SP/PIP2/PKC pathway (
Figure 2).
Table 2.
Research on the effects of ethanol and its derivatives in vitro.
Figure 2. TRPV1 inflammation nociceptor activation and ethanol-induced neuroinflammation. Key: CGRP: calcitonin gene-related peptide; PKC: protein kinase C; NGF: nerve growth factor; LDCV: large dense-core vesicle; SP: substance P; PIP2: phosphatidylinositol biphosphate; DAG: diacylglycerol.
2.2. Toll-Like Receptor R4
Several studies report ethanol-induced responses through the TLR4 receptor, an in vitro neuroinflammatory trigger
[39][48][66,75]. Another study found that after 30 min of ethanol treatment, NF-κB and AP-1 were upregulated in cultured rat cortical astrocytes. There was also the upregulation of iNOS and COX-2, culminating in the production of IL-1RI and IL-1β
[35][62] (see
Table 2). In subsequent research, it was confirmed that lipid rafts were vital to the recruitment of IL-1R1 and TLR4 receptors and triggered their endocytosis by cavesomes
[34][61]. This stimulated their downstream signaling and ultimately induced the upregulation of IL-1β. The other related inflammation factors in the TLR4 receptor signal transduction pathway, such as TNF-α, MCP-1, and NF-κB, were also upregulated in a series of cell tests
[28][49][50][55,76,77].
In addition to ethanol, EtG and acetaldehyde have also been suggested to elicit AIHs via TRPV1 or TLR4
[39][48][66,75]. A study observed that TRPV1 and TLR4 mRNA levels were upregulated by ethanol in wild-type mice
[16][43]. TLR4
−/− mutant mice showed higher TRPV1 mRNA expression relative to the control group in response to ethanol treatment. Thus, multiple lines of evidence imply that TRPV1 and TLR4 promote AIHs through neuroinflammation.
A study proposed that lipopolysaccharide (LPS) affects neuroinflammation via TRPV1 and TLR4. The upregulation of TLR4 activates TRPV1 via PKCξ
[51][78]. Other studies that investigated this hypothesis reported that LPS
[52][79], histamine
[53][80], and other substances
[54][81] could sensitize TRPV1 through the upregulation of TLR4. Additionally, the AP-1 transcription factor was activated in a TRPV1-independent and -dependent manner with Ca
2+ inflow observed, linking both TRPV1 and TLR4 stimulation through AP-1
[55][82]. These results show a consistent relationship among ethanol, TRPV1, TLR4, and AIHs: ethanol first promotes TLR4, which induces the sensitization of TRPV1. Together they promote downstream inflammation factors (e.g., CGRP, SP, TNF-α, IL-1β, and PKC) that elicit AIHs with neuroinflammation.