Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Christopher D'Angelo.
The gut microbiome is increasingly being recognized as an important immunologic environment, with direct links to the host immune system. The scale of the gut microbiome’s genomic repertoire extends the capacity of its host’s genome by providing additional metabolic output, and the close communication between gut microbiota and mucosal immune cells provides a continued opportunity for immune education. The relationship between the gut microbiome and the host immune system has important implications for oncologic disease, including lymphoma, a malignancy derived from within the immune system itself.
- lymphoma and gut microbiome
- gut microbiome and lymphomagenesis
- microbial interventions in lymphoma
1. Introduction
The gut microbial genome is confined within the human intestine and is estimated to be 150 times larger than the entire human genome, much of this belonging to bacteria [1,2][1][2]. Hence, the gut microbiome is often referred to as a second human genome [1]. The gut microbiome is a collective term for an ecosystem formed by different microorganisms colonizing the human gastrointestinal tract [3]. Microbial dysbiosis is a broad term defined as the loss of “health-promoting” microbes that are commensal to the gut, and/or the deleterious presence of pathogenic ones [4]. This imbalance has been found to be associated with myriad conditions such as chronic inflammatory states, which include inflammatory bowel diseases, infections, and immune dysregulation, resulting in various immune-mediated diseases and neoplastic conditions, including hematologic malignancies [5,6][5][6].
In recent years, the study of gut microbiota has received significant attention from researchers attempting to uncover ties between the microbiome and human health. The balance of gut microbiota has been shown to be associated with many cancers and studies are continuing to evaluate their interactions with different cancer treatments. For example, researchers have demonstrated the potentially crucial role of gut microbiota in influencing colon cancer development and progression, through unfavorable microbial community states in the human gut. Specifically, bacteria such as Bacteroides fragilis [7,8][7][8] and Fusobacterium nucleatum [9] are known to induce a pro-inflammatory state in the colon that could potentially enhance oncogenic proliferation in the mucosal microenvironment. There is also a growing body of evidence correlating the role of gut microbiota and hematologic malignancies. The contributions or role of the microbiome may differ with respect to its separate impact on disease pathogenesis and therapy.
2. Microbiome Environments and Lymphomagenesis
Animal models provide a useful tool in determining the intricate relationship between the gut microbiome and health and disease; in particular, the importance of the microbiota in immune development and composition, as well as its role in carcinogenesis [16][10]. Investigators have demonstrated its significant contribution to disease progression and the eradication of organisms such as Staphylococcus aureus, with improved outcomes in cutaneous T-cell lymphoma (CTCL) [17][11]. Tegla et al. also demonstrated the increased colonization of Staphylococcus spp. via CTCL microbiome analysis, when compared with healthy controls. This suggests a potential role of the dominance of S. aureus, leading to direct enhanced antigen presentation, enhanced clonal expansion of T cells, and the upregulation of pro-inflammatory cytokines, contributing to disease progression in CTCL [17][11]. Broad-spectrum antibiotics used in the treatment of advanced-stage CTCL lesions were associated with tumor regression and a reduction in the fraction of malignant T cells, suggesting that the microbiota can contribute to lymphoma pathogenesis [18][12]. Primary lymphomas that recapitulate the features of gastric mucosa-associated lymphoid tissue lymphoma (gastric MALT lymphoma) are known to arise in the stomach [19,20][13][14]. Epidemiologically, gastric MALT lymphoma is closely associated with the infection of a specific bacteria, Helicobacter pylori [21][15]. While H. pylori is present in approximately 50% of the world’s population [21][15], the incidence of gastric MALT lymphoma is low, suggesting that there are specific mechanisms by which H. pylori invades the gastric mucosa and, subsequently, evades the host immune system. The first study to investigate and further elucidate this link was conducted by Wotherspoon et al. In this study, the investigators showed that H. pylori infection significantly increased the risk of gastric MALT lymphoma, due to the vast majority of these patients being infected with H. pylori [19][13]. Furthermore, this study observed that the lymphoid follicles in these H. pylori-infected individuals developed into lymphoid tissues that were morphologically identical to gastric MALT lymphoma [19][13]. In 1993, Wotherspoon et al. further characterized the role of H. pylori in gastric MALT lymphoma using six patients, all of whom showed histological and genetic evidence of gastric MALT lymphoma. Remarkably, treatment with antibiotics led to the eradication of H. pylori infection and the subsequent regression of the lymphoma in the majority of the study population [22][16]. Since then, H. pylori infection and the different mechanisms contributing to the genesis of gastric MALT lymphoma have been revealed. Tumor-infiltrating T cells that are stimulated by H. pylori antigens have been shown to enhance the growth of B cells in gastric MALT lymphoma [23][17]. Additionally, another study showed that antigen-dependent development and the subsequent progression of gastric MALT lymphoma may be triggered by the expression of the CD40 ligand, in concert with Th2 cytokines, IL-4, and IL-10, by activated T cells [24][18]. These studies highlight the intricate communication between microbes, T cells, and B cells in the pathogenesis of MALT lymphoid malignancies and suggest that the balance of immune-activating and immune-suppressing cell populations and their signals contributes to the unique features in MALT lymphoma, including susceptibility to antibiotic-driven strategies. Specific tumor-infiltrating T cells and B cell receptor (BCR) signaling also play a role in the pathogenesis of gastric MALT lymphoma. In a study conducted by Craig et al., investigators used a murine model of H. felis-induced gastric MALT lymphoma to show that the development of MALT lymphoma requires both BCR signaling, via the poly-reactivation of tumor-derived immunoglobulins (Igs) with self-antigens, and tumor-infiltrating CD4+ T cells [25][19]. Moreover, most of these CD4+ T cells were FOXP3+ regulatory T cells that were being recruited by the tumor cells via chemokines such as CCL17 and CCL22 [25][19]. The in vivo inhibition of FOXP3+ regulatory T cells indeed resulted in the regression of gastric MALT lymphoma [25][19]. In line with this finding, Garcia et al. showed a higher FOXP3+/CD3+ cell ratio in H. pylori-positive gastric MALT lymphoma than in H. pylori-negative gastric MALT lymphoma [26][20]. Additionally, the expression of CD86, a co-stimulatory molecule that activates B cells to proliferate and produce IgG in B-cell lymphoma, was shown to be significantly associated with the sensitivity of H. pylori-dependent gastric MALT lymphoma to H. pylori eradication [27][21]. As illustrated in Figure 1, we demonstrate the intertwined relationship involving the gut microbiome, lymphomagenesis, lymphoma-directed therapies, and microbial interventions, details of which will be given later in this article.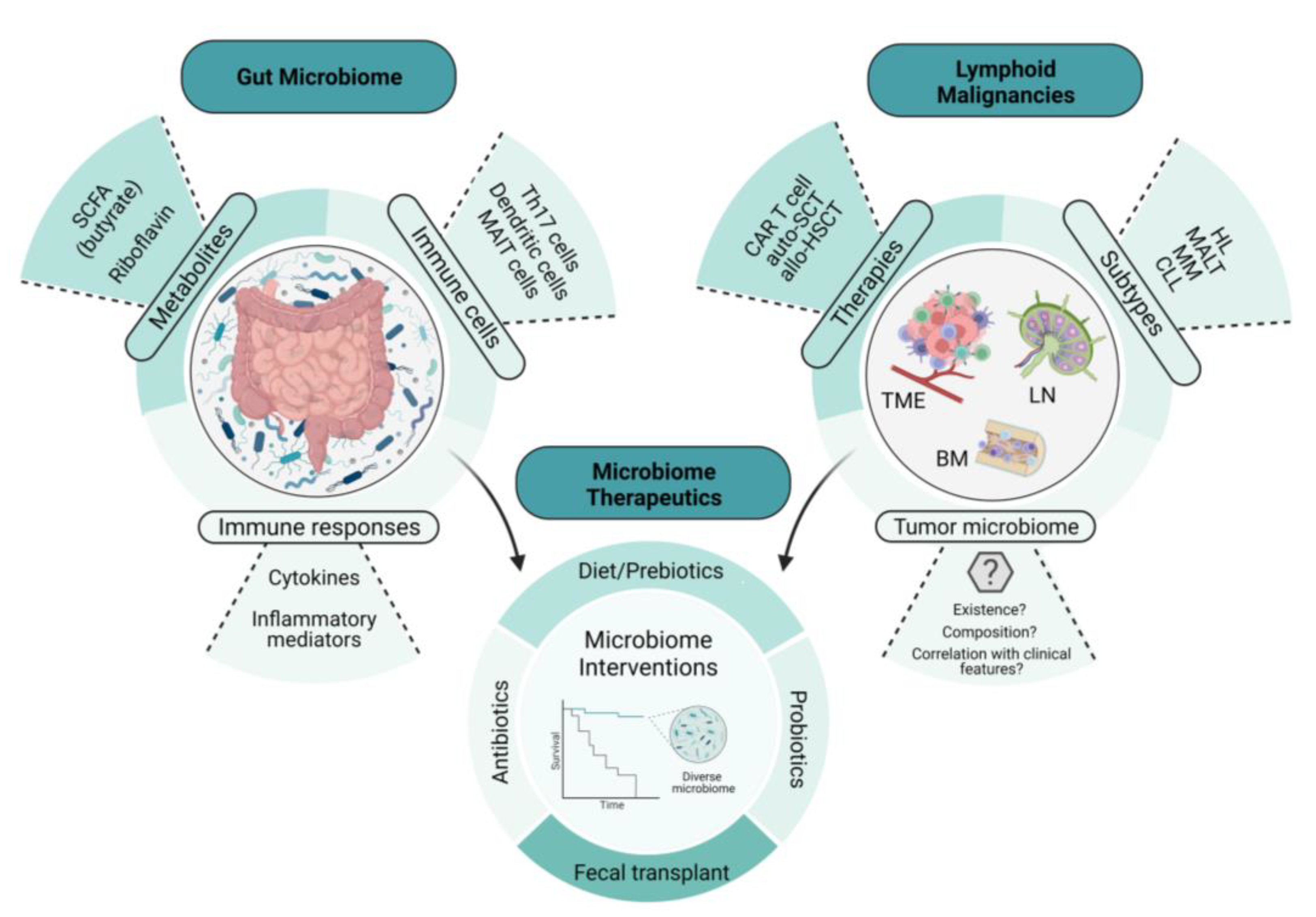
Figure 1. The intertwined relationship between the gut microbiome, lymphomagenesis, lymphoma-directed therapies, and microbiome-directed interventions. This illustration depicts the relationship between the gut microbiome, lymphoid malignancies, and microbiome therapeutics. The examples of microbiome therapeutics that are provided demonstrate how these approaches change key features and readouts of the gut microbiome. These changes in the microbiome not only influence lymphoid malignancies directly but also affect the therapies used to treat them, including immune cellular therapies such as CAR T-cell therapy, potentially through the emerging role of the tumor microbiome. SCFA, short-chain fatty acid; CAR T cell, chimeric antigen receptor T cell; auto-SCT, autologous stem-cell transplant; allo-HSCT, allogeneic hematopoietic stem-cell transplant; HL, Hodgkin’s lymphoma; MALT, mucosa-associated lymphoid tissue; MM, multiple myeloma; CLL, chronic lymphocytic leukemia; TME, tumor microenvironment; BM, bone marrow; LN, lymph node.
References
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65.
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836.
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379.
- Severyn, C.J.; Brewster, R.; Andermann, T.M. Microbiota modification in hematology: Still at the bench or ready for the bedside? Blood Adv. 2019, 3, 3461–3472.
- Frick, J.-S.; Autenrieth, I.B. The gut microflora and its variety of roles in health and disease. Between Pathog. Commens. 2012, 358, 273–289.
- Savage, D.C. Microbial Ecology of the Gastrointestinal Tract. Annu. Rev. Microbiol. 1977, 31, 107–133.
- Purcell, R.V.; Pearson, J.; Aitchison, A.; Dixon, L.; Frizelle, F.A.; Keenan, J.I. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS ONE 2017, 12, e0171602.
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022.
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215.
- Yamamoto, M.L.; Schiestl, R.H. Intestinal Microbiome and Lymphoma Development. Cancer J. 2014, 20, 190–194.
- Tegla, C.A.; Herrera, A.M.; Seffens, A.M.; Fanok, M.H.; Dean, G.; Kawaoka, J.; Laird, M.E.; Fulmer, Y.; Willerslev-Olsen, A.; Hymes, K.B.; et al. Skin Associated Staphylococcus Aureus Contributes to Disease Progression in CTCL. Blood 2019, 134, 659.
- Lindahl, L.M.; Willerslev-Olsen, A.; Gjerdrum, L.M.R.; Nielsen, P.R.; Blumel, E.; Rittig, A.H.; Celis, P.; Herpers, B.; Becker, J.C.; Stausbol-Gron, B.; et al. Antibiotics inhibit tumor and disease activity in cutaneous T-cell lymphoma. Blood 2019, 134, 1072–1083.
- Wotherspoon, A.C.; Ortiz-Hidalgo, C.; Falzon, M.R.; Isaacson, P.G. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991, 338, 1175–1176.
- Farinha, P.; Gascoyne, R.D. Helicobacter pylori and MALT lymphoma. Gastroenterology 2005, 128, 1579–1605.
- Kuo, S.H.; Wu, M.S.; Yeh, K.H.; Lin, C.W.; Hsu, P.N.; Chen, L.T.; Cheng, A.L. Novel Insights of Lymphomagenesis of Helicobacter pylori-Dependent Gastric Mucosa-Associated Lymphoid Tissue Lymphoma. Cancers 2019, 11, 547.
- Wotherspoon, A.C.; Doglioni, C.; Diss, T.C.; Pan, L.; Moschini, A.; de Boni, M.; Isaacson, P.G. Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993, 342, 575–577.
- Hussell, T.; Isaacson, P.G.; Crabtree, J.E.; Spencer, J. The response of cells from low-grade B-cell gastric lymphomas of mucosa-associated lymphoid tissue to Helicobacter pylori. Lancet 1993, 342, 571–574.
- Greiner, A.; Knorr, C.; Qin, Y.; Sebald, W.; Schimpl, A.; Banchereau, J.; Muller-Hermelink, H.K. Low-grade B cell lymphomas of mucosa-associated lymphoid tissue (MALT-type) require CD40-mediated signaling and Th2-type cytokines for in vitro growth and differentiation. Am. J. Pathol. 1997, 150, 1583–1593.
- Craig, V.J.; Cogliatti, S.B.; Arnold, I.; Gerke, C.; Balandat, J.E.; Wundisch, T.; Muller, A. B-cell receptor signaling and CD40 ligand-independent T cell help cooperate in Helicobacter-induced MALT lymphomagenesis. Leukemia 2010, 24, 1186–1196.
- Garcia, M.; Bellosillo, B.; Sanchez-Gonzalez, B.; Garcia-Payarols, F.; Seoane, A.; Ferrer, A.M.; Gimeno, E.; Barranco, L.E.; Torner, A.; Sole, F.; et al. Study of regulatory T-cells in patients with gastric malt lymphoma: Influence on treatment response and outcome. PLoS ONE 2012, 7, e51681.
- de Jong, D.; Vyth-Dreese, F.; Dellemijn, T.; Verra, N.; Ruskone-Fourmestraux, A.; Lavergne-Slove, A.; Hart, G.; Boot, H. Histological and immunological parameters to predict treatment outcome of Helicobacter pylori eradication in low-grade gastric MALT lymphoma. J. Pathol. 2001, 193, 318–324.
More