Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Yihang Zhang.
An important contributor to the development of idiopathic pulmonary fibrosis (IPF) is the alteration of the intracellular homeostasis of alveolar epithelial cells, which are mainly composed of alveolar type I epithelial cells (AT1), alveolar type II epithelial cells (AT2), as well as abnormal basaloid cells, resulting in aberrant epithelial repair, myofibroblast activation, and increased extracellular matrix deposition to form lung fibrosis
- idiopathic pulmonary fibrosis
- alveolar epithelial cells
- niche cells
1. Alveolar Epithelial Type I Cells
AT1 is morphologically flat and covers more than 95% of the surface area of the lung epithelium, which together with the basement membrane, alveolar wall capillaries form the air–blood barrier and are mainly responsible for gas exchange. Efficient gas exchange relies on the intrinsic ion and fluid transport functions of AT1 cells [15][1]. In addition, AT1 expresses pro-inflammatory receptors, including toll-like receptor 4 (TLR4) and receptors for advanced glycation end-products (RAGE) involved in innate immunity [16,17][2][3]. In pulmonary fibrosis, AT1 has received relatively little attention because AT1 is generally considered to be a terminally differentiated cell that does not have the functions of proliferation and differentiation itself [18,19][4][5]. However, in contrast to AT2, which is more responsive to hyperoxic injury, AT1 cells rapidly shed after exposure to bleomycin (BLM)-induced lung injury, and are completely lost during the subsequent fibrotic process [20][6]. There are several lines of evidence supporting that AT1 cells may participate in pulmonary fibrosis. RAGE, which is selectively expressed in AT1 cells and acts as a regulator of inflammation, was found to either promote fibrosis or controversially repress the progression of lung fibrosis [21,22,23,24][7][8][9][10]. Caveolin-1, which encode a scaffold protein of caveolae and is mainly present in AT1, could directly inhibit TGF-β signaling and frequently lost its expression in pulmonary fibrosis [25,26][11][12]. Recently, the role of AT1 in alveologenesis and alveolar regeneration is beginning to receive increasing attention. Studies have shown that Hopx+Igfbp2-AT1 cells can be transdifferentiated into AT2 cells to participate in alveolar injury repair [27,28,29,30][13][14][15][16]. These studies have changed the view that AT1 is a terminally differentiated cell. Therefore, the role of AT1 in pulmonary fibrosis needs to be further elucidated (Figure 1).
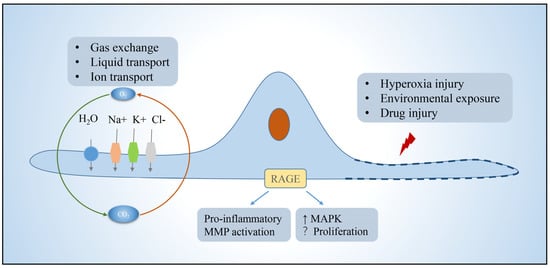
Figure 1. Function of alveolar type I epithelial cells. AT1 in the lung is mainly responsible for gas exchange, ion, and fluid transport, and can participate in cellular immunity. AT1 achieves fluid transport via aquaporins, ion transport via the cystic fibrosis transmembrane regulator and ATPases, and proinflammatory, pathway activating, immune, and other functions via a variety of protein receptors such as rage, which is localized to the basement membrane. Function is impaired or cells die when exposed to environmental insults. Abbreviations: RAGE, receptors for advanced glycation end-products; MMP, matrix metalloproteinases; MAPK, mitogen-activated protein kinase.
2. Alveolar Epithelial Type II Cells
AT2, defined as alveolar stem cells, has the ability of self-renewal and differentiation, which produces pulmonary surfactant, reduces alveolar epithelial surface tension, and prevents alveolar collapse. After alveolar epithelial cell damage, AT2 cells can rapidly proliferate and differentiate into AT1 cells, thereby maintaining the integrity of alveolar epithelial cells, which is necessary for maintaining alveolar homeostasis and promoting alveolar regeneration [31][17]. Notably, a subset of AT2 cells expressing the transcriptional target of Wnt signaling, Axin2, has been shown to play a leading role in alveolar regeneration and repair, and can be rapidly mobilized, self-renewed, and differentiated into AT1 cells after injury [32][18]. Disruption of protein homeostasis, telomere damage, mitochondrial dysfunction, and epigenetic changes lead to AT2 dysfunction, manifested as impaired stem cell function, apoptosis, senescence, and pro-fibrotic signaling, which are closely related to the development of IPF and which we will review in detail in this section.
During the initial injury phase, activated alveolar epithelial cells and recruited inflammatory cells release potent pro-fibrotic growth factors (e.g., TGF-β, tumor necrosis factor α, platelet-derived growth factor PDGF) that induce injury and fibrogenesis [33][19]. These growth factors, especially TGFβ, are involved in the damage and apoptosis of alveolar epithelial cells, induction of epithelial–mesenchymal transformation (EMT) of alveolar epithelial cells, activation and invasion of fibroblasts, and ECM deposition [34,35][20][21]. Human, mouse, and rat alveolar epithelial cells display a pattern of mesenchymal gene expression with the involvement of transcription factors when stimulated with TGFβ in vitro [34][20]. Sulforaphane (SFN) was found to enhance antioxidant capacity, reverse TGF-β-induced interstitial changes, return cells to epithelioid morphology, and exhibit significant antifibrotic effects on IPF patients, TGF-β-treated cell lines, and BLm-induced fibrosis in mice [36,37][22][23]. AT2 cells play a central role in the activation of TGFβ, which may be self-sustained by elevated tension in the fibrotic lung [6][24]. Cell division cycle 42 (Cdc42) acts on the polymerization of actin in AT2 cells, and its deletion causes a sustained increase in mechanical tension in AT2 cells, leading to activation of the TGF-β pathway, driving the comprehensive development of fibrosis from the periphery to center, with low Cdc42 expression in IPF patient samples [35,38][21][25]. Activation of TGF-β in AT2 induces the production of fibrosis, which can be alleviated by inhibition of TGF-β transduction.
2.1. Protein Homeostasis Destruction
Stemness maintenance of stem cells depends on tightly controlled protein homeostasis [39][26]. The role of the endoplasmic reticulum (ER) in vivo is to facilitate the folding and transport of proteins to ensure the quality of proteins required for cellular homeostasis. Protein misfolding triggers ER stress, leading to unfolded protein response (UPR), and persistent ER stress causes cellular dysfunction and affects stem cell fate decisions [40,41][27][28]. The study found that during pulmonary fibrosis, ER stress marker aggregation and UPR activation existed in AT2 cells. In BLM-injured mice, AT2 cells also exhibited ER stress, and the ER stress activator tunicamycin exacerbates pulmonary fibrosis [42,43][29][30]. Deletion of glucose-regulated protein 78 (GRP78), a key regulator of ER homeostasis, causes AT2 cells to undergo ER stress, apoptosis, senescence, impaired stemness, and TGF-β/Smad signaling activation; moreover, GRP78 is severely deleted in AT2 cells of IPF patients [44][31]. Wang further found that microcystin-leucine arginine (LR) can reduce TGF-β/Smad signaling, regulate macrophage polarization, block EMT and myofibroblast differentiation, and bind to GRP78 to inhibit the UPR signaling pathway, which contributes to alleviating pulmonary fibrosis [45][32]. ER stress and activation of UPR can cause AT2 dysfunction and further promote the progression of pulmonary fibrosis, which can be alleviated by inhibiting its activation.
2.2. Mitochondrial Damage
Mitochondria possess their own genetic material and genetic system. In addition to providing energy for cells, mitochondria are also involved in processes such as cell proliferation and differentiation, cell information transmission, and cell apoptosis, and have the ability to regulate cell growth and the cell cycle. Impaired mitochondrial phagocytosis, DNA damage, increased reactive oxygen species, growth disturbance, dysfunction, and disturbance of homeostasis in AT2 cells all induce ER stress and programmed cell death in AT2 cells, leading to the development of fibrosis [46,47][33][34]. Mitochondria were damaged and deformed in AT2 cells in IPF compared with control lungs, showing enlargement, swelling, and cristae rupture [48,49][35][36]. PTEN-induced putative kinase 1 (Pink1), an enzyme that promotes phagolysis of damaged mitochondria, is downregulated in IPF patients, and the degree of lung fibrosis is increased in Pink1-deficient mice, which is associated with reduced mitophagy, accumulation of malformed mitochondria, ER stress, and increased AT2 apoptotic senescence [43][30]. Activating transcription factor 3 (Aft3) is highly expressed in fibrotic and aging lungs [48][35]. Conditional deletion of Aft3 in AT2 protects mice from pulmonary fibrosis [50][37]. Mitochondrial (mt) DNA base excision repair enzyme, 8-oxoguanine-DNA glycosylase 1 (mtOGG1), can prevent mtDNA damage and apoptosis in epithelial cells. It was found that OGG1-deficient mice had increased pulmonary fibrosis, and IPF patients had increased lung mtDNA damage. mtOGG1 overexpression protected mitochondrial DNA integrity in epithelial cells and weakened cell apoptosis [51][38]. Phosphoglycerate mutase family member 5 (PGAM5), an important regulator of mitochondrial homeostasis in pulmonary fibrosis, impairs mitochondrial integrity at the functional and structural levels. Ganzleben et al. found that PGAM5-deficient mice and human epithelial cell fibrosis was significantly reduced [52][39]. These data suggest that improved mitochondrial homeostasis has protective effects on AT2 cells and provides protection against pulmonary fibrosis [53][40].
2.3. Telomere Shortening
Telomeres maintain the proliferative potential of stem and progenitor cells by providing a telomerase-dependent repeat expansion mechanism that protects chromosome ends from replicative loss [54][41]. Telomere shortening impairs stem cell function and tissue regeneration [55][42]. Telomere shortening in the alveolar epithelium is a common factor in disease progression, and telomere length measured in AT2 is uniformly reduced in IPF [56][43]. Telomeres are shorter in cells in fibrotic areas compared to non-fibrotic areas in IPF lungs [57][44]. IPF progression has been shown to be associated with mutations in the telomerase reverse transcriptase family genes, telomerase RNA component (TERC) and telomerase reverse transcriptase (TERT), which regulate telomere length and function [58][45]. Dysfunctions such as telomere shortening resulting from gene mutations lead to cell cycle arrest, AT2 senescence, and impaired renewal capacity, which are risk factors for the development of IPF [5,59,60][46][47][48]. Recent studies have found that POT1 p. (L259S) is defective in binding telomere protrusion, nuclear accumulation, negative regulation of telomerase, and lagging chain maintenance. Heterozygous mutations in this gene in IPF patients exhibit telomere loss, lagging strand defects, DNA damage, and cellular senescence, and mutations in this gene are thought to be a pathogenic driver of IPF [61][49]. Overexpression of E3 ubiquitin-protein ligase FBW7 (F-box and WD40 repeat domain-containing 7, also termed FBXW7) inhibits the expression of telomere capping enzyme tripeptidyl peptidase 1 (TPP1), leading to shortening of telomeres, senescence of AT2 cells, and promotion of the occurrence and development of IPF [62][50]. Krüppel-like factor 4 (KLF4), a protein transcription factor, is involved in a variety of cellular processes and plays an important role in the maintenance of cellular stemness. The study found that the expression of KLF4 and TERT in IPF patients and fibrosis mouse model AT2 is decreased, while the overexpression of KLF4 can increase the expression of TERT and telomerase activity [63][51]. Therefore, maintaining the stemness of AT2 stem cells by regulating telomere length and function may be a new way to alleviate the development of fibrosis.
3. Abnormal Basaloid Cells
In recent years, studies using single-cell sequencing technology and lineage tracing technology have found that there is a previously unknown alveolar differentiation intermediate (ADI, also called basaloid cells) population in the differentiation progress of AT2 cells to AT1 cells during the repair of alveolar epidermal damage in mice [64,65][52][53]. ADI expresses keratin 17/8 (KRT17/KRT8) but does not express the classic AT2 marker Surfactant Protein C (SFTPC) and AT1 markers advanced glycosylation end-product specific receptor (AGER), podoplanin (PDPN), and can further differentiate into mature AT1 cells [64][52]. In vivo and in vitro functional experiments confirmed that intervention in the ADI-specific regulatory network can promote or inhibit its differentiation into AT1 cells. Surprisingly, the population of basaloid cells is significantly increased in IPF patients and highly enriched in areas of severe fibrosis [7][54]. Furthermore, human basaloid cell populations can be transdifferentiated into keratin 5 (KRT5)-positive basal stem cells and their progeny, which largely explains why patients with IPF exhibit an alveolar-bronchialized phenotype [66][55]. In vitro organoid and in vivo xenograft experiments further confirmed that human basaloid cell populations can differentiate into normal alveolar epithelial cells or transdifferentiate into basal-like cells to promote fibrosis in immunodeficient mice [66][55] (Figure 2).
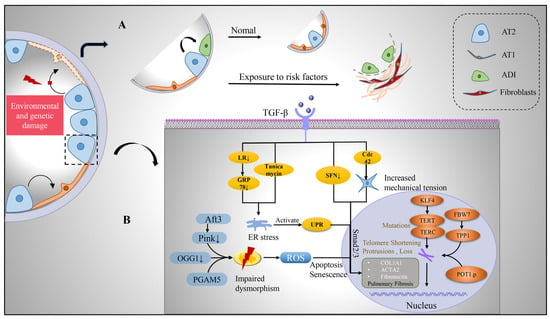
Figure 2. Transition of alveolar epithelium and intrinsic pathological mechanism of alveolar type II cells after injury. (A) Upon environmental and genetic insults to the alveolar epithelium, AT2 with stem cell functions undergoes self-renewal and differentiation. During its differentiation into AT1, a transitional cell morphology, the ADI, is generated, which is enriched in IPF lungs. (B) However, after AT2 is damaged, pro-fibrosis factor TGF-β is released, which can affect the expression of pro-fibrosis gene, produce endoplasmic reticulum stress, produce UPR, increase cell mechanical pressure, and activate TGFβ-Smad2/3 pathway. Mitochondria in damaged AT2 were damaged and deformed, with enlargement and swelling, ridge breakage, increased reactive oxygen species, inducing apoptosis and senescence of cells. TERT and TERC mutated and telomere shortened and lost under the influence of intracellular genes, which jointly promoted the development of pulmonary fibrosis. Abbreviations: AT1, alveolar type I epithelial cells; AT2, alveolar type II epithelial cells; ADI, alveolar differentiation intermediate; TGF-β, transforming growth factor-β; LR, leucine arginine; ER, endoplasmic reticulum; UPR, unfolded protein response; ROS, reactive oxygen species; GRP78, glucose-regulated protein 78; SFN, sulforaphane; KLF4, Krüppel-like factor 4; TERT, telomerase reverse transcriptase; TERC, telomerase RNA component; FBW7, F-box and WD40 repeat domain-containing 7; TPP1, tripeptidyl peptidase 1; Aft3, Pink1, PTEN-induced putative kinase 1; OGG1, 8-oxoguanine-DNA glycosylase 1; PGAM5, phosphoglycerate mutase family member 5.
References
- Dobbs, L.G.; Johnson, D.M.; Vanderbilt, J.; Allen, L.; Gonzalez, R. The great big alveolar TI cell: Evolving concepts and paradigms. Cell. Physiol. Biochem. 2010, 25, 55–62.
- Wong, M.H.; Chapin, O.C.; Johnson, M.D. LPS-stimulated cytokine production in type I cells is modulated by the renin-angiotensin system. Am. J. Respir. Cell Mol. Biol. 2012, 46, 641–650.
- Stogsdill, M.P.; Stogsdill, J.A.; Bodine, B.G.; Fredrickson, A.C.; Sefcik, T.L.; Wood, T.T.; Kasteler, S.D.; Reynolds, P.R. Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 49, 128–134.
- Weibel, E.R. On the tricks alveolar epithelial cells play to make a good lung. Am. J. Respir. Crit. Care Med. 2015, 191, 504–513.
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp. Mol. Pathol. 1975, 22, 142–150.
- Song, M.J.; Davidovich, N.; Lawrence, G.G.; Margulies, S.S. Superoxide mediates tight junction complex dissociation in cyclically stretched lung slices. J. Biomech. 2016, 49, 1330–1335.
- Queisser, M.A.; Kouri, F.M.; Konigshoff, M.; Wygrecka, U.; Schubert, M.; Eickelberg, O.; Preissnel, K.T. Loss of RAGE in pulmonary fibrosis: Molecular relations to functional changes in pulmonary cell types. Am. J. Respir. Cell Mol. Biol. 2008, 39, 337–345.
- Shirasawa, M.; Fujiwara, N.; Hirabayashi, S.; Ohno, H.; Iida, J.; Makita, K.; Hata, Y. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 2004, 9, 165–174.
- Ding, H.; Ji, X.; Chen, R.; Ma, T.; Tang, Z.; Fen, Y.; Cai, H. Antifibrotic properties of receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Pulm. Pharmacol. Ther. 2015, 35, 34–41.
- He, M.; Kubo, H.; Ishizawa, K.; Hegab, A.E.; Yamamoto, Y.; Yamamoto, H.; Yamaya, M. The role of the receptor for advanced glycation end-products in lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1427–L1436.
- Shivshankar, P.; Brampton, C.; Miyasato, S.; Kasper, M.; Thannickal, V.J.; Le Saux, C.J. Caveolin-1 deficiency protects from pulmonary fibrosis by modulating epithelial cell senescence in mice. Am. J. Respir. Cell Mol. Biol. 2012, 47, 28–36.
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906.
- Wang, Y.; Tang, Z.; Huang, H.; Li, J.; Wang, Z.; Yu, Y.; Zhang, C.; Li, J.; Dai, H.; Wang, F.; et al. Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 2018, 115, 2407–2412.
- Zepp, J.A.; Morley, M.P.; Loebel, C.; Kremp, M.M.; Chaudhry, F.N.; Basil, M.C.; Leach, J.P.; Liberti, D.C.; Niethamer, T.K.; Ying, Y.; et al. Genomic, epigenomic, and biophysical cues controlling the emergence of the lung alveolus. Science 2021, 371, eabc3172.
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.J.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat. Commun. 2015, 6, 6727.
- Penkala, I.J.; Liberti, D.C.; Pankin, J.; Sivakumar, A.; Kremp, M.M.; Jayachandran, S.; Katzen, J.; Leach, J.P.; Windmueller, R.; Stolz, K.; et al. Age-dependent alveolar epithelial plasticity orchestrates lung homeostasis and regeneration. Cell Stem Cell 2021, 28, 1775–1789.e5.
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036.
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123.
- Zhang, C.; Zhu, X.; Hua, Y.; Zhao, Q.; Wang, K.; Zhen, L.; Wang, G.; Lu, J.; Luo, A.; Cho, C.W.; et al. YY1 mediates TGF-beta1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir. Res. 2019, 20, 249.
- Fernandez, I.E.; Eickelberg, O. New cellular and molecular mechanisms of lung injury and fibrosis in idiopathic pulmonary fibrosis. Lancet 2012, 380, 680–688.
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17.
- Kyung, S.Y.; Kim, D.Y.; Yoon, J.Y.; Son, E.S.; Kim, Y.J.; Park, J.W.; Jeong, S.H. Sulforaphane attenuates pulmonary fibrosis by inhibiting the epithelial-mesenchymal transition. BMC Pharmacol. Toxicol. 2018, 19, 13.
- Artaud-Macari, E.; Goven, D.; Brayer, S.; Hamimi, A.; Besnard, V.; Marchal-Somme, J.; Ali, Z.E.; Crestani, B.; Kerdine-Romer, S.; Boutten, A.; et al. Nuclear factor erythroid 2-related factor 2 nuclear translocation induces myofibroblastic dedifferentiation in idiopathic pulmonary fibrosis. Antioxid. Redox Signal. 2013, 18, 66–79.
- Xu, Y.; Mizuno, T.; Sridharan, A.; Du, Y.; Guo, M.; Tang, J.; Wikenheiser-Brokamp, K.A.; Perl, A.-K.T.; Funari, V.A.; Gokey, J.; et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 2016, 1, e90558.
- Shochet, G.E.; Israeli-Shani, L.; Kains, I.; Wand, O.; Shitrit, D. MiR-608 overexpression in idiopathic pulmonary fibrosis (IPF). BMC Pulm. Med. 2021, 21, 1.
- García-Prat, L.; Sousa-Victor, P.; Muñoz-Cánoves, P. Proteostatic and Metabolic Control of Stemness. Cell Stem Cell 2017, 20, 593–608.
- Delbrel, E.; Soumare, A.; Naguez, A.; Label, R.; Bernard, O.; Bruhat, A.; Fafournoux, P.; Tremblais, G.; Marchant, D.; Gille, T.; et al. HIF-1alpha triggers ER stress and CHOP-mediated apoptosis in alveolar epithelial cells, a key event in pulmonary fibrosis. Sci. Rep. 2018, 8, 17939.
- Delbrel, E.; Uzunhan, Y.; Soumare, A.; Gille, T.; Marchant, D.; Planes, C.; Boncoeur, E. ER Stress is Involved in Epithelial-To-Mesenchymal Transition of Alveolar Epithelial Cells Exposed to a Hypoxic Microenvironment. Int. J. Mol. Sci. 2019, 20, 1299.
- Lawson, W.E.; Cheng, D.-S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567.
- Bueno, M.; Lai, Y.-C.; Romero, Y.; Brands, J.; Croix, C.M.S.; Kamga, C.; Corey, C.; Herazo-Maya, J.D.; Sembrat, J.; Lee, J.; et al. PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis. J. Clin. Investig. 2015, 125, 521–538.
- Borok, Z.; Horie, M.; Flodby, P.; Wang, H.; Liu, Y.; Ganesh, S.; Firth, A.L.; Minoo, P.; Li, C.; Beers, M.F.; et al. Grp78 Loss in Epithelial Progenitors Reveals an Age-linked Role for Endoplasmic Reticulum Stress in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 198–211.
- Wang, J.; Xu, L.; Xiang, Z.; Ren, Y.; Zheng, X.; Zhao, Q.; Zhou, Q.; Zhou, Y.; Xu, L.; Wang, Y. Microcystin-LR ameliorates pulmonary fibrosis via modulating CD206(+) M2-like macrophage polarization. Cell Death Dis. 2020, 11, 136.
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820.
- Mora, A.L.; Bueno, M.; Rojas, M. Mitochondria in the spotlight of aging and idiopathic pulmonary fibrosis. J. Clin. Investig. 2017, 127, 405–414.
- Jaeger, V.K.; Lebrecht, D.; Nicholson, A.G.; Wells, A.; Bhayani, H.; Gazdhar, A.; Tamm, M.; Venhoff, N.; Geiser, T.; Walker, U.A. Mitochondrial DNA mutations and respiratory chain dysfunction in idiopathic and connective tissue disease-related lung fibrosis. Sci. Rep. 2019, 9, 5500.
- Patel, A.S.; Song, J.W.; Chi, S.G.; Mizumura, K.; Osorio, J.C.; Shi, Y.; El-Chemaly, S.; Lee, C.G.; Rosas, I.O.; Elias, J.A.; et al. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS ONE 2015, 10, e0121246.
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; Croix, C.S.; Sembrat, J.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720.
- Kim, S.-J.; Cheresh, P.; Jablonski, R.P.; Rachek, L.; Yeldandi, A.; Piseaux-Aillon, R.; Ciesielski, M.J.; Ridge, K.M.; Gottardi, C.J.; Lam, A.P.; et al. Mitochondrial 8-oxoguanine DNA glycosylase mitigates alveolar epithelial cell PINK1 deficiency, mitochondrial DNA damage, apoptosis, and lung fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1084–L1096.
- Ganzleben, I.; He, G.-W.; Günther, C.; Prigge, E.-S.; Richter, K.; Rieker, R.J.; Mougiakakos, D.; Neurath, M.F.; Becker, C. PGAM5 is a key driver of mitochondrial dysfunction in experimental lung fibrosis. Cell. Mol. Life Sci. 2019, 76, 4783–4794.
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; Werneck-De-Castro, J.P.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49.
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309.
- Piñeiro-Hermida, S.; Autilio, C.; Martínez, P.; Bosch, F.; Pérez-Gil, J.; Blasco, M.A. Telomerase treatment prevents lung profibrotic pathologies associated with physiological aging. J. Cell Biol. 2020, 219.
- Lee, J.S.; La, J.; Aziz, S.; Dobrinskikh, E.; Brownell, R.; Jones, K.D.; Achtar-Zadeh, N.; Green, G.; Elicker, B.M.; Golden, J.A.; et al. Molecular markers of telomere dysfunction and senescence are common findings in the usual interstitial pneumonia pattern of lung fibrosis. Histopathology 2021, 79, 67–76.
- Snetselaar, R.; Van Batenburg, A.A.; Van Oosterhout, M.F.M.; Kazemier, K.M.; Roothaan, S.M.; Peeters, T.; Van Der Vis, J.J.; Goldschmeding, R.; Grutters, J.C.; Van Moorsel, C.H.M. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE 2017, 12, e0189467.
- Diaz de Leon, A.; Cronkhite, J.T.; Katzenstein, A.L.; Godwin, J.D.; Raghu, G.; Glazer, C.S.; Rosenblatt, R.L.; Girod, C.E.; Garrity, E.R.; Xing, C.; et al. Telomere lengths, pulmonary fibrosis and telomerase (TERT) mutations. PLoS ONE 2010, 5, e10680.
- Barratt, S.; Creamer, A.; Hayton, C.; Chaudhuri, N. Idiopathic Pulmonary Fibrosis (IPF): An Overview. J. Clin. Med. 2018, 7, 201.
- Kropski, J.A.; Blackwell, T.S. Progress in Understanding and Treating Idiopathic Pulmonary Fibrosis. Annu. Rev. Med. 2019, 70, 211–224.
- Arish, N.; Petukhov, D.; Wallach-Dayan, S.B. The Role of Telomerase and Telomeres in Interstitial Lung Diseases: From Molecules to Clinical Implications. Int. J. Mol. Sci. 2019, 20, 2996.
- Kelich, J.; Aramburu, T.; van der Vis, J.J.; Showe, L.; Kossenkov, A.; van der Smagt, J.; Massink, M.; Schoemaker, A.; Hennekam, E.; Veltkamp, M.; et al. Telomere dysfunction implicates POT1 in patients with idiopathic pulmonary fibrosis. J. Exp. Med. 2022, 219.
- Wang, L.; Chen, R.; Li, G.; Wang, Z.; Liu, J.; Liang, Y. FBW7 Mediates Senescence and Pulmonary Fibrosis through Telomere Uncapping. Cell Metab. 2020, 32, 860–877.e9.
- Wang, H.; Xu, H.; Lyu, W.; Xu, Q.; Fan, S.; Chen, H.; Wang, D.; Chen, J.; Dai, J. KLF4 regulates TERT expression in alveolar epithelial cells in pulmonary fibrosis. Cell Death Dis. 2022, 13, 435.
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559.
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946.
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983.
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5(+) basal cells. Nat. Cell Biol. 2022, 24, 10–23.
More