Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Mariam Nikolaishvili.
Henoch–Schönlein purpura or IgA vasculitis is the most common type of pediatric vasculitis that may affect adults as well. It is classified as a type of small-vessel vasculitis. It can cause cutaneous and systemic symptoms with a minority of patients developing kidney failure.
- vasculitis
- IgA
- vessel
- virus
- covid 19
- infections
- purpura
- Henoch-Schonlein purpura
- arthritis
- joints
1. Introduction
Henoch–Schönlein purpura (HSP), also known as immunoglobulin A (IgA) vasculitis, is the most common form of systemic vasculitis in children, with a 20.4/100,000 population incidence rate [1]. Most cases occur in children between the ages of 2 and 8 years. Males are affected twice as frequently as females. HSP may infrequently affect adults as well. It is a small-vessel leukocytoclastic vasculitis caused by immune complex deposition, which may manifest as a systemic or single-organ restricted disease [2]. Commonly affected organs include the skin, kidney, gastrointestinal system, and joints [3]. Although there have been significant steps made toward understanding pathogenetic mechanisms, HSP etiology remains largely unknown. This condition would be induced by an abnormal inflammatory process deriving from immune reactions to various antigenic stimuli in genetically predisposed subjects [4]. The first-degree relatives of affected patients are at increased risk of developing this disease. A strong association with HLA class II alleles, specifically HLA-DRB1 alleles, has been identified. Then, peculiar immune complex deposits play a pivotal role in the pathogenesis with the resulting necrosis of the wall of small vessels.
HSP often occurs after bacterial or viral infections and is more frequent in the winter months. TResearche aim of this review is trs aim to make a critical appraisal of the possible relationship between viral infections, viral vaccines, and HSP.
2. Pathogenesis
The deposition of immune complexes containing IgA in the small vessels of the skin, the renal mesangium, and the additionally affected organs is the defining pathogenic aspect of HSP.
Human IgA displays a large heterogeneity as regards molecular forms and glycosylation [5] with two subclasses that are differentially distributed between the mucosal and circulatory compartments of the immune system. IgA1 and IgA2 are the two isotypes of IgA. IgA1 predominates in serum, while the percentages of IgA2 are higher in secretions. They may be generated in both monomeric and dimeric forms and are both highly glycosylated proteins. Their structure differs by the absence of a 13-amino acid sequence in the hinge region of the IgA2 molecule [6], which gives it a particular resistance against bacterial proteases and may explain the predominance of IgA2 in mucosal secretions.
The glycosylation and size of IgA1 appear to be crucial in promoting IgA1 molecule clearance [7]. Normal interactions between glycosylated IgA1 molecules and the hepatocyte-expressed asialoglycoprotein receptor (ASGP-R) result in the internalization and destruction of these molecules [8]. Patients with HSP, similarly to patients with IgA nephropathy, exhibit poorly galactosylated IgA1 O-glycoforms deficient in galactose and/or sialic acid [9]. However, it appears that an increase in the levels of poorly galactosylated IgA1 O-glycoforms is not sufficient in itself to develop HSP. Indeed, investigations involving the relatives of patients detected similar levels of poorly galactosylated IgA1 O-glycoforms without signs or history of HSP [8]. Consequently, it has been considered that a second, subsequent step may be required for the transition to the full phase of the disease. Aberrantly glycosylated IgA1 molecules expose N-acetylgalactosamine-containing neoepitopes, which may be recognized by glycan-specific IgG or IgA1 antibodies [10] (Figure 1 and Figure 2). The aberrant galactosylated IgA1 O-glycoforms might act either as autoantigens driving the formation of glycan-specific antibodies in genetically prone individuals or as antigens for cross-reactive antimicrobial antibodies. Evidence increasingly suggests that the next step is the formation of large circulating immune complexes prone to deposition in small vessels [11]. Indeed, soluble immune complexes, due to their increased size, are unable to pass through the space of Disse and connect with the asialoglycoprotein receptor (ASGP-R) on hepatocytes. They can pass through the larger fenestrae in the glomerular capillaries that lie directly above the mesangium. By the alternative complement pathway’s activation and the recruitment of inflammatory cells, these deposited complexes cause damage to glomeruli (Figure 3) [10,11,12][10][11][12]. IgA-containing immune complexes are discovered in patients’ serum, as well as the immune complexes containing C3 and IgA in the skin, intestines, and kidneys (Figure 4).
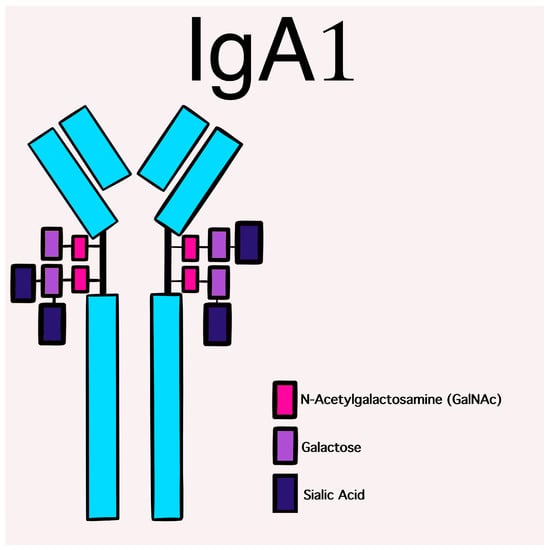
Figure 1. Schematic picture of a normal IgA1 containing GalNAc-galactose disaccharide and its mono- and di-sialylated forms in the hinge region of the heavy chains.
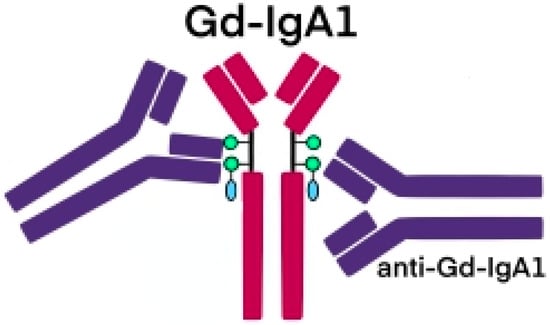
Figure 2. Schematic picture of abnormally glycosylated IgA1 exposing a novel antigenic determinant involving N-acetylgalactosamine (GalNAc), which may be recognized by naturally occurring specific antibodies.
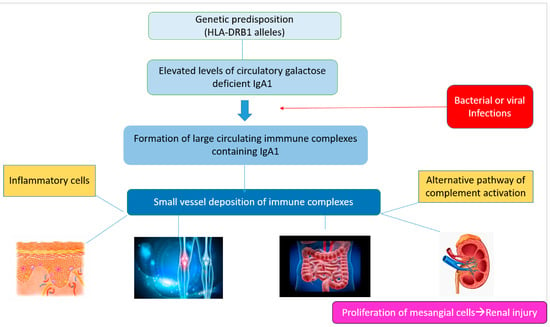
Figure 3.
Schematic representation of the main pathogenetic steps of IgA vasculitis.
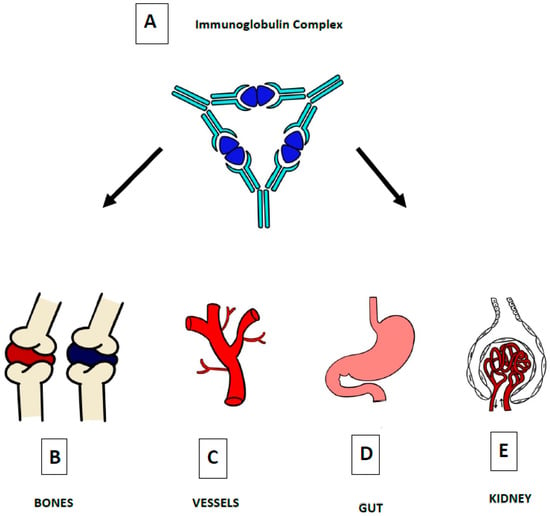
Figure 4. The defining pathogenic mechanism of HSP is due to IgA-containing immune complexes (A), which deposit in the small vessels of skin (C), joints (B), intestine (D), and kidneys (E).
3. Environment, Microbial, and Virus Infections
The etiology of HSP seems to be a combination of genetic predisposition, environmental factors, and infectious agents. Its etiology and pathogenesis remain not fully understood, but a number of factors, mainly infectious agents, drugs, and vaccines have been considered as possible triggers. HSP is more frequent in the autumn and winter months and is usually preceded by a wide variety of upper respiratory tract infections. However, community-based outbreaks of HSP have not been reported. Accordingly, there is enough evidence that only susceptible hosts may develop the disease. Indeed, familial clusters of HSP have been indicated, with siblings affected simultaneously or sequentially [13].
In addition to the pathological findings of IgA deposits on small-vessel walls, the occurrence of polymorphonuclear neutrophils infiltration around vessels, the elevation of IgA serum levels, and proinflammatory cytokines during the acute stage suggest that HSP is a specific immune-mediated entity induced by environmental factors, particularly infections [13]. Therefore, HSP is considered a post-infectious immune-mediated small-vessel vasculitis.
Various mechanisms have been proposed to link infections and HSP. Following infections, aberrant IgA1 could be able to recognize the GalNAc-containing (N-acetylgalactosamine-specific) molecules expressed on bacterial or viral structures and form circulating complexes. They would be deposited in the mesangium, inducing the activation of the mesangial cells, which would finally lead to renal damage. According to another pathogenic model, T-cell activation by microbes during respiratory infections could promote increased levels of the transforming growth factor (TGF)-β, which is able to induce an isotype switch of IgA and increased IgA serum levels [13]. Finally, mucosal infections could lead to the upregulation of IL-6 with the possible development of aberrant glycosylation of IgA1 [14].
Of all the pathogens linked to HSP, group A β-hemolytic streptococcus has been the most studied since it may be detected in up to 50% of individuals with acute HSP by serological testing or bacterial cultures [15]. However, multiple bacteria and viruses have been associated with the development of HSP.
References
- Gardner-Medwin, J.M.; Dolezalova, P.; Cummins, C.; Southwood, T.R. Incidence of Henoch-Schonlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins. Lancet 2002, 360, 1197–1202.
- Demirkesen, C. Approach to cutaneous vasculitides with special emphasis on small vessel vasculitis: Histopathology and direct immunofluorescence. Curr. Opin. Rheumatol. 2017, 29, 39–44.
- Hetland, L.; Susrud, K.; Lindahl, K.; Bygum, A. Henoch-Schönlein Purpura: A Literature Review. Acta Derm.-Venereol. 2017, 97, 1160–1166.
- Rigante, D.; Castellazzi, L.; Bosco, A.; Esposito, S. Is there a crossroad between infections, genetics, and Henoch–Schönlein purpura? Autoimmun. Rev. 2013, 12, 1016–1021.
- Monteiro, R.C. Role of IgA and IgA Fc Receptors in Inflammation. J. Clin. Immunol. 2009, 30, 1–9.
- Woof, J.M.; Kerr, M.A. The function of immunoglobulin A in immunity. J. Pathol. 2006, 208, 270–282.
- Person, T.; King, R.G.; Rizk, D.V.; Novak, J.; Green, T.J.; Reily, C. Cytokines and Production of Aberrantly O-Glycosylated IgA1, the Main Autoantigen in IgA Nephropathy. J. Interf. Cytokine Res. 2022, 42, 301–315.
- Kiryluk, K.; Moldoveanu, Z.; Sanders, J.T.; Eison, T.M.; Suzuki, H.; Julian, B.A.; Novak, J.; Gharavi, A.G.; Wyatt, R. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch–Schönlein purpura nephritis. Kidney Int. 2011, 80, 79–87.
- Trnka, P. Henoch-Schönlein purpura in children. J. Paediatr. Child Health 2013, 49, 995–1003.
- Novak, J.; Moldoveanu, Z.; Renfrow, M.B.; Yanagihara, T.; Suzuki, H.; Raska, M.; Hall, S.; Brown, R.; Huang, W.-Q.; Goepfert, A.; et al. IgA Nephropathy and Henoch-Schoenlein Purpura Nephritis: Aberrant Glycosylation of IgA1, Formation of IgA1-Containing Immune Complexes, and Activation of Mesangial Cells. IgA Nephrop. Today 2007, 157, 134–138.
- Boyd, J.K.; Barratt, J. Inherited IgA glycosylation pattern in IgA nephropathy and HSP nephritis: Where do we go next? Kidney Int. 2011, 80, 8–10.
- Novak, J.; Julian, B.A.; Tomana, M.; Mestecky, J. IgA Glycosylation and IgA Immune Complexes in the Pathogenesis of IgA Nephropathy. Semin. Nephrol. 2008, 28, 78–87.
- Yang, Y.-H.; Chuang, Y.-H.; Wang, L.-C.; Huang, H.-Y.; Gershwin, M.E.; Chiang, B.-L. The immunobiology of Henoch–Schönlein purpura. Autoimmun. Rev. 2008, 7, 179–184.
- Heineke, M.H.; Ballering, A.V.; Jamin, A.; Mkaddem, S.B.; Monteiro, R.C.; Van Egmond, M. New insights in the pathogenesis of immunoglobulin A vasculitis (Henoch-Schönlein purpura). Autoimmun. Rev. 2017, 16, 1246–1253.
- Saulsbury, F.T. Clinical update: Henoch-Schönlein purpura. Lancet 2007, 369, 976–978.
More