Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Hong Choi.
The adenosine triphosphate (ATP)-binding cassette transporter A1 (ABCA1) was identified by Luciani et al. over 36 years ago by PCR cloning and found to be located on chromosome 9q22-31. ABCA1 belongs to what was then a growing family of transmembrane proteins sharing many structural and functional similarities. ABCA1 was initially thought to be involved in the phagocytosis of apoptotic cells and to play a role in the regulation of the inflammatory response. Recent studies of structure-function relationships have shown that ABCA1 transports cholesterol and phospholipids across the plasma membrane to generate high-density lipoproteins (HDLs).
- ABCA1
- atherosclerosis
- cholesterol
- high-density lipoprotein
1. Brief History of ABCA1, Tangier Disease
The cholesterol molecule evolved through the eons of evolution to maintain proper cell function [1]. Cellular cholesterol homeostasis is exquisitely regulated by multiple, redundant systems that ensure a narrow concentration range [2]. Excess cholesterol leads to cell autophagy and apoptosis through the unfolded protein response [3]. Some cells, especially M2 macrophages, have developed the ability to esterify cholesterol and store it in cholesteryl ester vesicles for transport to the sites of cholesterol catabolism, such as the spleen in mammals. However, when this system is overwhelmed by an abundant availability of oxidized low-density lipoprotein cholesterol (LDL-C), it can lead to the formation of foam cells, one of the earliest pathological findings in atherosclerosis. The intracellular regulatory pathway for cholesterol has been reviewed elsewhere [2] and will not be further dealt with here. The removal of cholesterol from cells by apolipoprotein A-I (apoA-I) is another mechanism by which accumulated cellular cholesterol can egress [4].
An interesting patient phenotype was identified in a young boy from the island of Tangier in the Chesapeake Bay, Maryland in the early 1960s. The patient had enlarged tonsils, hepato-splenomegaly, and was referred to the burgeoning National Institutes of Health (NIH). On extensive phenotyping, he was found to have an absence of high-density lipoprotein cholesterol (HDL-C). This new disease, named Tangier disease, became the topic of intense research since. The cellular defect was later identified to be an inability of apoA-I to promote cellular cholesterol efflux [5]. Family studies located the defect on chromosome 9q22-31. Multiple lines of evidence indicated that Tangier disease was caused by a cellular defect in removing cholesterol by apoA-I. The term used, cellular cholesterol efflux defect, became the main focus of research into its etiology.
The adenosine triphosphate (ATP)-binding cassette transporter A1 (ABCA1) was identified by Luciani et al. over 36 years ago by PCR cloning and found to be located on chromosome 9q22-31 [6]. ABCA1 belongs to what was then a growing family of transmembrane proteins sharing many structural and functional similarities. ABCA1 was initially thought to be involved in the phagocytosis of apoptotic cells and to play a role in the regulation of the inflammatory response [7,8][7][8]. Several groups simultaneously reported that pathogenic variants in the ABCA1 gene were the molecular basis of Tangier disease, a recessive orphan disease characterized clinically by hepato-splenomegaly, enlarged tonsils, described pathologically as lipid-laden macrophages and lymphoid tissues, a progressive peripheral neuropathy, and represented biochemically by an absence of HDL-C in the blood [9,10,11,12,13][9][10][11][12][13].
2. The Structure and Function of ABCA1
The ATP-binding cassette (ABC) transporters are a superfamily of transmembrane proteins that utilize ATP to transport a variety of substrates across cellular membranes. The ABC-A subfamily comprises 12 members, ABCA1 to ABCA13, with ABCA11 representing a transcribed pseudogene and is mostly related with lipid trafficking [22][14]. ABCA1 drives cholesterol efflux by exporting cholesterol and phospholipids (PLs) across the plasma membrane (PM) [23][15] and is a full-size ABC transporter containing two extracellular domains (ECD1 and ECD2), two transmembrane domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 and NBD2), and two regulatory domains (RD1 and RD2) (Figure 1). The human ABCA1 protein is composed of 2261 amino acids and its structure was solved by single-particle cryo-electron microscopy (cryo-EM) in 2017 with resolutions of 3.9–4.1 Å (Figure 1A) [24][16]. The cryo-EM structure has revealed that ABCA1 is different from other ABC transporters in several features. First, ABC transporters typically form a cavity between the two TMDs to bind and translocate their substrates across membranes; however, the TMD1 and TMD2 in ABCA1 contact partially and do not form a sealed cavity, leading to exposure of the TMD side surfaces to the lipid bilayer. This suggests that ABCA1 may allow lateral entry of lipid substrates from the lipid bilayer. Second, it has been generally accepted that the alternation between inward- and outward-facing TMD conformations makes it possible for ABC exporters to translocate their substrates across membranes [23,25][15][17]. In the alternating access model, the TMD in ATP-free ABC exporters is opened toward the cytoplasm (inward-facing conformation), while the TMD in ATP-bound ABC exporters is opened toward the extracellular side (outward-facing conformation). As seen in Figure 1A,B, the TMD in ATP-free ABCA1 is in an outward-facing conformation, implying that the alternating access model may not be compatible with the ATP-dependent lipid flopping activity of ABCA1. Third, the ABC-A subfamily members, including ABCA1, are differentiated from most other ABC transporters due to the presence of large ECDs. The discovery of the ABCA1 structure has revealed that the distinguished ECDs in ABCA1 enclose a hydrophobic tunnel, ~60 Å in length. The PM is estimated to be 70–100 Å thick and composed primarily of a PL bilayer; therefore, the length of a single PL molecule may range from 35 to 50 Å, suggesting that the hydrophobic tunnel may serve as a passageway for the delivery of PLs. Based on the structural analysis, two ABCA1-mediated PL export mechanisms have been proposed (Figure 1A): (a) PLs in the inner leaflet of the PM enter into the interface between TMD1 and TMD2, subsequently they are flopped into the outward-facing transmembrane cavity, and then translocated into the hydrophobic tunnel [24][16]; (b) PLs in the outer leaflet of the PM diffuse into the outward-facing transmembrane cavity, and then they are translocated into the hydrophobic tunnel [26][18]. Both of the mechanisms are distinct from the mechanisms reported for the other ABC exporters. The cryo-EM structure of human ABCA4 was determined in 2021 with resolutions of 3.3–3.4 Å [27,28][19][20]. ABCA4 is one of the closest relatives of ABCA1, and the comparison of the two cryo-EM structures has exhibited that the general architecture of ABCA1 and ABCA4 in the ATP-free state is very similar, but that the cytoplasmic RDs of the two are substantially different [28,29][20][21]. The RDs in ABCA4 cross over the axis of symmetry to form a domain-swapped pair of latches, while the ABCA1 RDs seen in Figure 1A have been modeled to make simple contacts without domain-swapping [28,29][20][21]. At the amino acid level, the RDs in human ABCA1 and ABCA4 show 59% identity and 76% similarity. This high degree of sequence homology and the domain-swapped latches unveiled in the higher-resolution structure of ABCA4 suggest that the ABCA1 RDs may also form domain-swapped latches. Aller and Segrest carefully inspected the original cryo-EM density map of ABCA1 and have proposed an alternative structure of human ABCA1 that contains domain-swapped RDs (Figure 1B) [29][21]. These RDs function like latches, so they constrain the NBDs to hold them in close proximity even in the absence of ATP binding. The authors have speculated that the swapping of RDs may be conserved in the entire ABC-A subfamily, based on their amino acid sequence analysis [29][21]. This swapped architecture may also be important for the unique action mechanisms of the ABC-A subfamily in comparison to the other subfamilies. Sun and Li solved the cryo-EM structures of human ABCA1 in both ATP-free and ATP-bound states in 2022 at 3.1 Å resolution [30][22]. These structures have exhibited that the ABCA1 RDs form domain-swapped latches. More interestingly, these structures have revealed the presence of a sterol-like molecule in the ECDs, and the authors have compared ATP-free with ATP-bound structures to propose an ABCA1-mediated cholesterol export model (Figure 1C). First, in the ATP-free state, cholesterol is loaded into the inner leaflet space between the TMDs, where a putative cholesterol-binding site is located. Next, ATP binding to the NBDs pulls the NBDs closer together, stretching the TMDs, inducing conformational changes in the TMD-ECD interface, leading to a counter-clockwise rotation of the ECDs by approximately 30 degrees. The authors have suggested that the closure of the outward-facing transmembrane cavity during the TMD stretching, plus the significantly changed TMD-ECD interface conformation, may drive the translocation of cholesterol from the inner leaflet space into the hydrophobic tunnel. They speculated that the translocation of cholesterol from the outer membrane leaflet into the hydrophobic tunnel might also be possible. Finally, upon release of ADP (the product of ATP hydrolysis), the NBD, TMD, and ECD conformations return to their ATP-free states.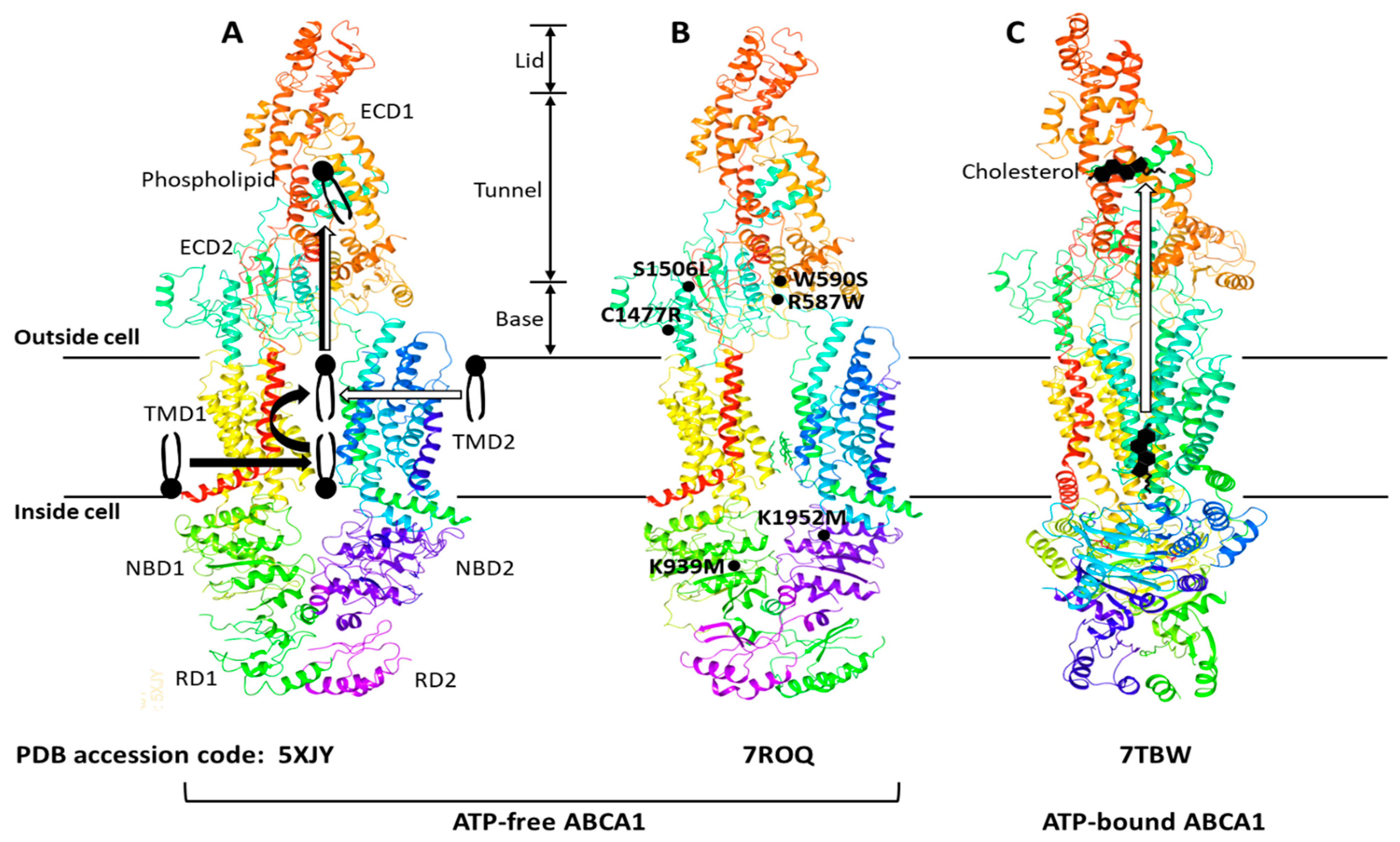
Figure 1. ABCA1 structures, proposed lipid transport mechanisms, and hypomorphic missense mutations. The ABCA1 comprises two extracellular domains (ECD1 and ECD2), two transmembrane domains (TMD1 and TMD2), two nucleotide-binding domains (NBD1 and NBD2), and two regulatory domains (RD1 and RD2). The ECD1 and ECD2 are intertwined, and the ECD can be subdivided into three parts—lid, hydrophobic tunnel, and base. The TMD1 and TMD2, each consisting of six transmembrane amphipathic α-helices, are separately folded. The RDs form a structural latch that may stabilize the NBDs and support effective cooperation between the two halves of ABCA1. Three ABCA1 structures—5XJY, 7ROQ, and 7TBW—were retrieved from the protein data bank (PDB) (https://www.rcsb.org) (accessed on 13 December 2022). Two different mechanisms of phospholipid export have been proposed with the initial structure 5XJY, and the mechanisms are depicted with thick black and white arrows in (A) [24,26][16][18]. The initial structure was revised to propose an alternative structure 7ROQ seen in (B) [29][21]. The RD1 and RD2 in (B) are swapped, and each RD forms a domain-swapped latch. The locations of representative hypomorphic missense mutations are shown in (B): R587W and S1506L variants are impaired in localization at the plasma membrane; W590S is defective in phospholipid translocation activity; C1477R reduces cellular apoA-I binding; K939M and K1952M disrupt ATPase activity [31][23]. A cholesterol export mechanism has been proposed with the 7TBW seen in (C) [30][22].
References
- Bloch, K.E. Evolutionary perfection of a small molecule. In Blondes in Venitian Paintings, the Nine-Banded Armadillo and Other Assays in Biochemistry; Yale University Press: New Haven, CT, USA, 1994; pp. 14–36. ISBN 0-300-05881.
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172.
- Tabas, I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ. Res. 2010, 107, 839–850.
- Oram, J.F. HDL apolipoproteins and ABCA1: Partners in the removal of excess cellular cholesterol. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 720–727.
- Mott, S.; Yu, L.; Marcil, M.; Boucher, B.; Rondeau, C.; Genest, J., Jr. Decreased cellular cholesterol efflux is a common cause of familial hypoalphalipoproteinemia: Role of the ABCA1 gene mutations. Atherosclerosis 2000, 152, 457–468.
- Luciani, M.F.; Denizot, F.; Savary, S.; Mattei, M.G.; Chimini, G. Cloning of two novel ABC transporters mapping on human chromosome 9. Genomics 1994, 21, 150–159.
- Becq, F.; Hamon, Y.; Bajetto, A.; Gola, M.; Verrier, B.; Chimini, G. ABC1, an ATP binding cassette transporter required for phagocytosis of apoptotic cells, generates a regulated anion flux after expression in Xenopus laevis oocytes. J. Biol. Chem. 1997, 272, 2695–2699.
- Schmitz, G.; Kaminski, W.E.; Porsch-Ozcurumez, M.; Klucken, J.; Orso, E.; Bodzioch, M.; Buchler, C.; Drobnik, W. ATP-binding cassette transporter A1 (ABCA1) in macrophages: A dual function in inflammation and lipid metabolism? Pathobiology 1999, 67, 236–240.
- Brooks-Wilson, A.; Marcil, M.; Clee, S.M.; Zhang, L.H.; Roomp, K.; van Dam, M.; Yu, L.; Brewer, C.; Collins, J.A.; Molhuizen, H.O.; et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat. Genet. 1999, 22, 336–345.
- Rust, S.; Rosier, M.; Funke, H.; Real, J.; Amoura, Z.; Piette, J.C.; Deleuze, J.F.; Brewer, H.B.; Duverger, N.; Denefle, P.; et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat. Genet. 1999, 22, 352–355.
- Bodzioch, M.; Orso, E.; Klucken, J.; Langmann, T.; Bottcher, A.; Diederich, W.; Drobnik, W.; Barlage, S.; Buchler, C.; Porsch-Ozcurumez, M.; et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat. Genet. 1999, 22, 347–351.
- Remaley, A.T.; Rust, S.; Rosier, M.; Knapper, C.; Naudin, L.; Broccardo, C.; Peterson, K.M.; Koch, C.; Arnould, I.; Prades, C.; et al. Human ATP-binding cassette transporter 1 (ABC1): Genomic organization and identification of the genetic defect in the original Tangier disease kindred. Proc. Natl. Acad. Sci. USA 1999, 96, 12685–12690.
- Hooper, A.J.; Hegele, R.A.; Burnett, J.R. Tangier disease: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 80–84.
- Albrecht, C.; Viturro, E. The ABCA subfamily–gene and protein structures, functions and associated hereditary diseases. Pflugers. Arch. 2007, 453, 581–589.
- Phillips, M.C. Is ABCA1 a lipid transfer protein? J. Lipid Res. 2018, 59, 749–763.
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239.e1210.
- Jardetzky, O. Simple allosteric model for membrane pumps. Nature 1966, 211, 969–970.
- Segrest, J.P.; Tang, C.; Song, H.D.; Jones, M.K.; Davidson, W.S.; Aller, S.G.; Heinecke, J.W. ABCA1 is an extracellular phospholipid translocase. Nat. Commun. 2022, 13, 4812.
- Xie, T.; Zhang, Z.; Fang, Q.; Du, B.; Gong, X. Structural basis of substrate recognition and translocation by human ABCA4. Nat. Commun. 2021, 12, 3853.
- Liu, F.; Lee, J.; Chen, J. Molecular structures of the eukaryotic retinal importer ABCA4. Elife 2021, 10, e63524.
- Aller, S.G.; Segrest, J.P. The regulatory domains of the lipid exporter ABCA1 form domain swapped latches. PLoS ONE 2022, 17, e0262746.
- Sun, Y.; Li, X. Cholesterol efflux mechanism revealed by structural analysis of human ABCA1 conformational states. Nat. Cardiovasc. Res. 2022, 1, 238–245.
- Wang, S.; Smith, J.D. ABCA1 and nascent HDL biogenesis. Biofactors 2014, 40, 547–554.
More