Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Giosuè Costa and Version 3 by Conner Chen.
Multiple myeloma (MM) is an aggressive and incurable disease for most patients, characterized by periods of treatment, remission and relapse. The introduction of new classes of drugs, such as proteasome inhibitors (PIs), has improved survival outcomes in these patient populations. The proteasome is the core of the ubiquitin–proteasome system (UPS), a complex and conserved pathway involved in the control of multiple cellular processes, including cell cycle control, transcription, DNA damage repair, protein quality control and antigen presentation.
- proteasome
- natural compounds
- proteasome inhibitors
1. Introduction
Multiple myeloma (MM) is the second most common hematologic malignancy worldwide, characterized by the proliferation of terminally differentiated antibody-producing plasma cells (PCs) [1][2][1,2]. Although MM has intrinsic genetic heterogeneity [3], a common feature of malignant clones is the production of elevated amounts of immunoglobulins [1], ultimately leading to organ dysfunctions such as hypercalcemia, renal insufficiency, anemia and bone disease (overall referred to as CRAB criteria) [4][5][4,5]. The bone marrow microenvironment (BMM) has been shown to play a significant role in MM pathogenesis triggering PC survival, proliferation and drug resistance [1][2][1,2]; moreover, genetic complexity, defined by chromothripsis and hyperdiploidy, along with copy number variations and single-nucleotide polymorphisms, account for the early evolution from asymptomatic stages (e.g., monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM)) to overt disease [6]. Additional alterations, including aberrant DNA or histone methylation [7] and microRNA (miRNA) dysregulation [8][9][8,9], may contribute to disease progression.
The ubiquitin–proteasome system (UPS) is crucial homeostatic machinery for protein degradation; it constantly regulates protein turnover, thus affecting various cellular functions, spanning from cell cycle regulation to survival, apoptosis, metabolism and protein quality control [10]. Since PCs secrete high amounts of immunoglobulins, they are strictly dependent on the UPS machinery and are highly reliant on the deregulation of protein degradation. Moreover, due to the constitutive activation of the nuclear factor kappa B (NF-κB) signaling pathway, malignant PCs are more sensitive to PI than healthy ones [11]. In fact, the anti-MM activity of proteasome inhibitors (PIs) was originally ascribed to the inhibition of oncogenic NF-κB through the blockage of the degradation of its negative regulator IκBα [12]. However, additional processes whose targeting contributes to the anti-tumor effects of PIs have subsequently been identified, including the reversal of cell cycle aberrations, apoptosis induction, endoplasmic reticulum stress, angiogenesis and DNA repair, as well as the reactivation of hypermethylated tumor-suppressor genes [12][13][14][15][12,13,14,15].
The exquisite sensitivity of MM cells to PIs, along with the design of successful clinical protocols, have led to their approval for the treatment of MM patients, with three compounds currently being used in clinics [16]. PIs are true milestones for the treatment of MM and other hematologic malignancies (e.g., mantle cell lymphoma), and are currently being investigated for other diseases. The first approved PI was bortezomib, a slowly reversible inhibitor of the β5 catalytic proteasomal subunit, followed by carfilzomib, an irreversible inhibitor of the β5 site, and by the first oral PI, ixazomib [2].
Natural products have always played a key role in drug discovery, especially for cancer and infectious diseases. In oncology, several drugs derived from Nature have been approved, and are widely used in clinics, including paclitaxel, romidepsin, vincristine and vinblastine [17][18][19][20][17,18,19,20].
Importantly, marine-derived natural compounds isolated from sponges, mollusks, cyanobacteria, corals and tunicates have been clinically approved for MM by the US and Australian FDA as well as the European Medicines Agency (EMA), such as belantamab mafodotin and plitidepsin, while others are under clinical trial or extensive preclinical assessment [21].
Additionally, naturally derived compounds with PI activity have been isolated from various sources, and are currently emerging as potential anti-cancer drugs in a wide variety of cancers, including MM [22].
2. Proteasome Structure and Function
By ruling protein degradation, the UPS represents a fundamental intracellular system whose components act in a highly coordinated manner through different steps, such as the polyubiquitylation, deubiquitylation and degradation of the target protein [16][23][16,23]. Thanks to the coordinated activities of this system, all the damaged or misfolded proteins, or the proteins which are no longer needed, are correctly destroyed [16]. The proteasome is the central core of this system. The eukaryotic 26S proteasome is a large (1500–2000 kDa) multi-subunit complex that degrades most cellular proteins under physiologic conditions. It is a multicatalytic proteinase complex able to bind, deubiquitylate and unfold its substrates prior to completing their degradation. The proteasome is predominately located in the cytosol and nucleus, and its functions are not limited to the maintenance of proteostasis—it is involved in a wide array of biological processes, including cell differentiation and proliferation, DNA repair and apoptosis, regulation of gene expression and response to stress [24][25][24,25]. Alternative forms of the proteasome, such as the immunoproteasome and thymoproteasome, are involved in routine proteolytic functions, antigen processing and T-cell selection [16]. The 26S proteasome is a dynamic ATP-dependent macromolecular machine, comprising two subcomplexes: the 19S regulatory particle (RP) and the 20S core particle (CP). Either one or two 19S RPs can attach to a single 20S core particle to form the 26S proteasome (19S-20S) (Figure 1A) or the 30S proteasome (19S-20S-19S), respectively. However, both conformations in the literature are referred to as “26S” (Figure 1B) [26][27][28][26,27,28].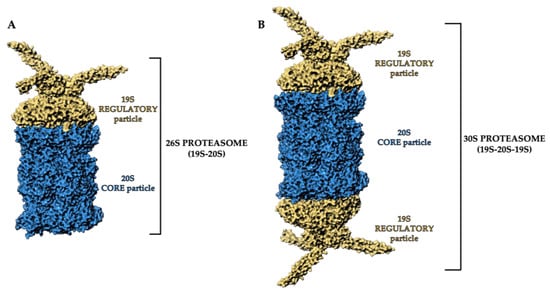
Figure 1. Three-dimensional representation of (A) 26S proteasome and (B) 30S proteasome structure. The 19S regulatory particles and the 20S core particle are shown as yellow and blue areas, respectively. Atomic coordinates were obtained from PDB model 5MP9 [29]; the figure was built by means of Maestro graphical interface [30].
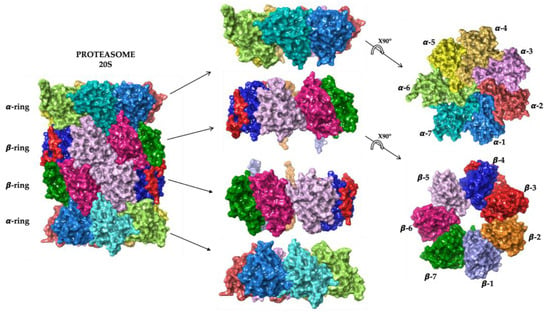
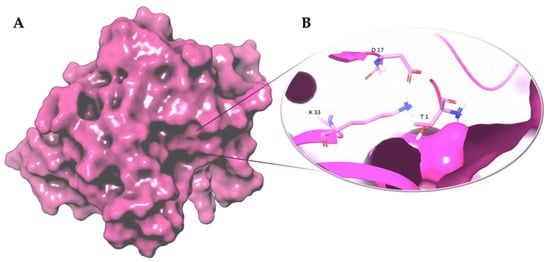
Figure 3. Three-dimensional representation of (A) proteasome chymotrypsin-like site (β5); (B) Thr-Lys-Asp catalytic triad of the chymotrypsin-like site (β5). The β5 subunit is represented as magenta surface and the conserved residues are shown as magenta carbon sticks. Atomic coordinates were obtained from PDB model 5LF7 [33]; the figure was built by means of Maestro graphical interface [30].
Approved and Investigational PIs for MM Treatment
PIs are classified according to their chemical structure and their mechanism of action. Covalent inhibitors are generally electrophilic and react with the catalytic gamma-hydroxyl of Thr1 in the active sites to reversibly or irreversibly inhibit the proteasome depending on the strength of the chemical bond. Based on their chemical structure, PIs can be classified in the following classes: peptide aldehydes, peptide boronates, epoxomicin and epoxyketones, lactacystin, β-lactone and vinyl sulfones. Similar to most protease inhibitors, several PIs are short peptides designed to fit into the substrate binding site of the catalytic subunit. Although the proteasome has three types of catalytic sites, full inhibition of all of them is not required to significantly affect protein degradation. In fact, while β5 inhibition leads to significantly reduced protein breakdown, specific β1 or β2 inhibition does not have any significant effect. Consequently, most PIs act by targeting the β5 site, although they can often have some lesser activity against β1 and/or β2 [37]. To date, three FDA-approved proteasome inhibitors, namely bortezomib (Velcade®), carfilzomib (Kyprolis®) and ixazomib (Ninlaro®), are in clinical use, and several drugs are under development (Figure 4).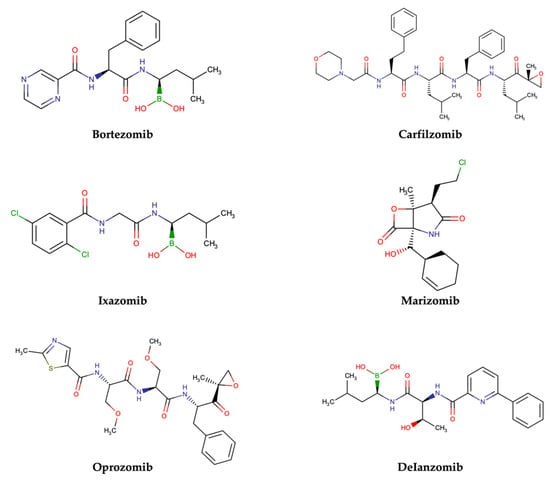
Figure 4.
Two-dimensional structures of approved and investigational PIs.
-
Bortezomib
-
Carfilzomib
-
Ixazomib
-
Marizomib
-
Oprozomib
Name, pharmacokinetics and pharmacodynamics characteristics of proteasome inhibitors.
-
Delanzomib
Table 1.
| Compound | Pharmacophoric Moiety | Binding Kinetics | Proteasome Subunit |
|---|---|---|---|
| Bortezomib | Boronate | Reversible | β5 > β1 |
| Carfilzomib | Epoxyketone | Irreversible | β5 |
| Ixazomib | Boronate | Reversible | β5 > β1 |
| Marizomib | β-lactone | Irreversible | β5 > β1 > β2 |
| Oprozomib | Epoxyketone | Irreversible | β5 |
| Delanzomib | Boronate | Reversible | β5 > β1 |