A huge number of factors are involved in controlling energy homeostasis, and nutrients are known to exert crucial epigenetic effects on metabolism.
Under CR and fasting, the production of KBs also increases and they can be used as energy fuel by the brain as an alternative to glucose. Furthermore, recent evidence has shed new light on the role of KBs in controlling protein acetylation, underlining the close link between KBs and SIRTs.
2. The Epigenetics Regulation at a Glance
Epigenetics are defined as a set of modifications in gene expression without any changes in the gene sequence
[19]. Although alterations in gene function can induce heritable phenotypic changes, this heritability across generations is less well established
[20]. In any case, the alteration of gene expression patterns governed by epigenetics can result in diseases
[21][22][21,22].
Hundreds of post-translational modifications (PTMs) have been found that can regulate cell differentiation, cell-specific gene expression, parental imprinting, X-chromosome inactivation, and genomic stability and structure. These include acetylation and deacetylation, methylation, and demethylation, phosphorylation, ubiquitination on the amino terminal or histone tail, and noncoding RNAs
[19]. Several chromatin-modifying enzymes are responsible for adding and removing histone modifications contributing to the complex epigenetic process
[19][23][19,23]. Moreover, the study of epigenetics is further complicated by the fact that the epigenome is tissue-specific, or even cell-specific, and that it changes over time
[24]. Furthermore, numerous metabolic cofactors are required to support the catalytic activity of enzymes, and the availability and variation of the dietary-introduced metabolites influence epigenetic regulation
[25][26][25,26].
The intake of macronutrients and, more importantly, the amount, the timing, and the CR, while contributing to changes in cellular metabolism and the availability of NAD+ are crucial in inducing epigenomic adaptations. In patients with nutritional derangements resulting in metabolic disorders, such as obesity and diabetes mellitus, an association with DNA hypermethylation has been observed
[27]. Interestingly, the KD positively modifies the redox state of the cell and modulates the activity of NAD+-dependent enzymes in deacetylation processes
[28]. However, the majority of this evidence is derived from blood cells, limiting their interpretation in a clinical setting
[24]. The study of the functional synergism between KBs and the NAD+-dependent deacetylase SIRT1—both involved in beneficial cellular metabolic processes—becomes of primary importance for understanding the epigenetic evolution of the KD, and of nutritional intervention in general (
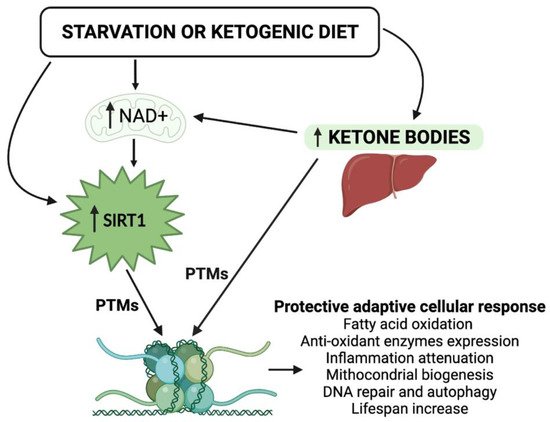
Figure 1). In view of the steady increase in metabolic disorders, including obesity, type 2 diabetes mellitus (T2DM), and cardiovascular diseases,
rwe
searchers strongly believe that any nutritional therapeutic approach—with a particular focus on the KDs—should be adopted in order to reduce the burden of these diseases. Moreover, the in-depth study of epigenetic modifications induced by KBs and SIRT1 is of particular interest today, because it could represent an additional tool in the management of metabolic dysfunctions.
Figure 1. Schematic representation of the epigenetic activity of ketone bodies and SIRT1 in response to starvation or to the ketogenic diet. Synergistically, KBs and SIRT1 target both histone and nonhistone proteins and alter cellular metabolic programs. NAD, nicotinamide adenine dinucleotide; PTMs, post-translational modifications.
3. Ketone Bodies and Epigenetic Regulation
β-OHB, AcAc, and acetone are a set of fuel molecules that serve as an alternative energy source to glucose in the event of carbohydrate restriction. Other than being produced by the liver from fatty acids during periods of fasting or prolonged/intense physical activity, the endogenous production of KBs is promoted by consuming the KD, which consists of the intake of a very low amount of carbohydrates (generally, <50 g per day). In contrast to the pathological state of ketoacidosis, which can occur in decompensated diabetes, the nutritional induction of mild ketonemia has been proven to be beneficial in animal models, leading to improved metabolic profiles and neurological responses, as well as a prolonged lifespan
[4][29][4,29]. Feeding elderly mice with a cyclic KD without reducing calory intake can decrease midlife mortality and prevent memory decline
[30]. Furthermore, the administration of the KD to aged male mice preserves motor function and muscle mass and extends longevity
[31].
In humans, the KD was initially used as a therapeutic option for its anticonvulsant effects in children with refractory epilepsy
[32]; then, a very low carbohydrate ketogenic diet (VLCKD) was introduced for the treatment of obesity and its complications, such as insulin-resistance, type 2 diabetes
[33], nonalcoholic fatty liver diseases (NAFLD)
[34], and obstructive sleep apnea syndrome
[35]. Moreover, apart from weight loss, the KD is widely used for the improvement of cardiovascular
[28], rheumatological
[36], neuronal
[37], and, currently, cancer diseases
[38][39][38,39].
Despite their beneficial use for several diseases, the systemic impact of the KDs is still only partially understood. Similar to what occurs during CR, the shift from carbohydrate to fat metabolism seen during the KD, and the subsequent oxidation of fatty acid, which increases the formation of KBs, appears to be responsible for the positive metabolic effects due to the intrinsic epigenetic function of KBs
[40]. Indeed, β-OHB action is associated with protection against energy depletion, oxidative stress, inflammation, and apoptosis, and this could represent the downstream effect of KBs on antioxidant pathways in counteracting senescence
[41].
The main histone PTMs induced by KBs are DNA methylation and histone phosphorylation, ubiquitination, and acetylation, which appear to be the key epigenetic mechanisms of β-OHB activity to modulate inflammation
[42].
β-OHB-mediated hyperacetylation was observed under CR. This epigenetic activity modulates specific induction in gene expression, such as, for example, the Forkhead Box O3 A (FOXO3A), a family of proteins functioning as sensors of the insulin signaling pathway and regulators of longevity, which, in turn, regulates DNA transcription
[30][43][30,43].
In addition to this direct mechanism, KBs ensure histone hyperacetylation through the inhibition of some deacetylase activities. Previous studies reported β-OHB as an endogenous inhibitor of class I histone deacetylase (HDACs) by competing for the catalytic site with butyrate, the structurally similar canonical HDAC inhibitor
[9][43][44][9,43,44]. Shimazu et al. showed that β-OHB induces HDAC1, HDAC3, and HDAC4 (classes I and IIa) inhibition in renal cells driving the upregulation of FOXO3A transcription factor network genes, including the antioxidant catalase, the mitochondrial superoxide dismutase (SOD), and the metallothionein 2
[44].
In contrast, some findings did not confirm the prominent function of β-OHB as a histone deacetylase inhibitor
[45], as well as the multilevel lysine hyperacetylation being questioned
[42][46][42,46]. Even if the discussion on the HDAC inhibitory potential of β-OHB is still open, its overall effect on chromatin and mitochondrial protein acetylation could be attributable to the severe increase in intracellular acetyl-CoA and the cell potential intended as the NAD+/NADH ratio.
It should now be pointed out that the deacetylases-inhibiting activity of KBs may seem to be in contrast with the deacetylation activity carried out by SIRT1. To clarify this aspect, it is necessary to focus on the fact that KBs specifically inhibit histone deacetylases belonging to class I and IIa, and that this is not in conflict with SIRTs, which belong to the class III family of deacetylases. However, since fasting and nutritional ketosis act differently on different classes of HDACs, how these potentially opposing activities coordinate remains an open question and deserves further study.
Furthermore, the increase in NAD+ levels following KD activity is a factor available for the SIRT1 NAD+dependent activation, thus, further promoting deacetylation
[47]. Interestingly, the metabolic shift towards fat oxidation and ketogenesis during starvation or during the KD is associated with initial mitochondrial stress characterized by increased levels of reactive oxygen species (ROS) and increased ratios of NAD+/NADH and adenosine monophosphate/adenosine triphosphate (AMP/ATP), as well as adenosine monophosphate/adenosine diphosphate (AMP/ADP). However, in the long-term, these “danger signals” provoke a protective and adaptive (hormetic) cellular response via the activation of SIRT1 and AMP-activated protein kinase (AMPK), and the consequences of the initial moderate metabolic stress include the upregulation of antioxidative and anti-inflammatory activities and an improved mitochondrial function
[47].
As mentioned above, DNA methylation also represents a well know KB epigenetic action. Several studies have highlighted the antiepileptic efficacy of KBs, which, through altering DNA methylation, changes the expression of epileptogenic genes
[48][49][50][48,49,50]. Some authors indicate KDs as exerting their disease-modifying effects through an adenosine-dependent epigenetic mechanism
[51]. In general, similarly to hyperacetylation, the histone methylation status seems to be related to the pool of acetyl-CoA, which, together with glycine, is required for the synthesis of S-adenosylmethionine (SAM). Interestingly, a classical KD deprived of threonine (and, therefore, deficient in SAM, glycine, and acetyl-CoA) may paradoxically exacerbate seizures, as reported in epileptic rodent models
[52]. In line with these findings, other studies showed that through DNA hypomethylation, KBs impact cardiac function. Indeed, hypomethylation affects the inflammatory functions of leukocytes related to cardiovascular risk through the modulation of adhesion/migration and soluble molecules
[28], and global DNA hypomethylation has been observed in atherosclerotic lesions of mice, rabbits, and humans, and in hypertensive patients
[28]. The hypomethylating activity of KBs may also have potential implications for the amelioration of metabolic diseases, in which DNA hypermethylation is frequently observed, as previously reported
[27].
Recent findings suggest that β-OHB coordinates cellular functions via a novel epigenetic modification termed the β-hydroxybutyrylation, that integrates classic DNA methylation and PTMs, and includes specific histone lysines (up to 44 domains susceptible to this activity) and cellular proteins such as p53
[52]. As previously reported in renal cells and rodent models, exposure to high levels of β-OHB induces the β-hydroxybutyrylation of lysines 9 and 14 (H3K9/K14) in histone 3, whose increase leads directly to the upregulation of genes involved in hunger-sensitive metabolic networks that represent a fundamental mechanism of energetic and metabolic adaptation
[53].
Finally, KBs also affect microRNAs (miRNAs). In volunteers treated with the KD, miRNAs directly targeted genes linked to nutrient metabolism, such as the mechanistic target of rapamycin (mTOR), peroxisome proliferator-activated receptors (PPARs), insulin, and cytokines, suggesting that the KD may modify miRNA expression, leading to the reduction in inflammatory interleukins such as IL-1β and IL-6, thus, mitigating neuroinflammation
[3].
Ketone metabolism can affect the activity of SIRTs through the regulation of NAD+ availability, as well as that of other substrates. The epigenetic role of nutritional ketosis and, more generally, the relationship between macronutrients and gene expression need to be further explored.
4. SIRT1 and Epigenetic Regulation
SIRT1-related epigenetic mechanisms have been specifically investigated thanks to robust studies previously performed on yeast, which identified the
Sir2 gene as being responsible for lifespan extension
[54]. The role of
Sir2 in the aging process has also been confirmed in higher organisms, such as Caenorhabditis elegans and Drosophila melanogaster
[55][56][55,56], and several findings have led to the recognition of the
Sir2-aging mechanism as an evolutionarily conserved system. Mammalian SIRT1 has been shown to exhibit similar activity to that of yeast
Sir2, except for substrate deacetylase, which not only includes histones, but also key transcription factors such as p53, with an effect on tumorigenesis and maintaining normal cell growth, as well as FOXO
[57][58][57,58].
Mainly located in the cell nucleus, in both prokaryotes and eukaryotes, SIRT1 can preferentially deacetylate lysine histone residues (H3K9 and H4K16)
[59][60][59,60], as confirmed in SIRT1-deficient mouse embryonic fibroblasts characterized by hyperacetylated histones, heterochromatin formation, and transcription repression
[61][62][61,62]. Expressed throughout all mammalian somatic and germ tissues, SIRT1 is responsible for aging-dependent global transcriptional changes caused by chromatin modification
[11]. During starvation or a CR, SIRT1 regulates the expression of molecules involved in the response to fasting such as PPAR-γ, PPARγ-coactivator1-α (PGC1-α), FOXO, uncoupling proteins (UCPs), etc. Specifically, by regulating the expression of PPAR-γ, which exerts a pleiotropic role on inflammation, metabolism, endothelial function, oxidative stress, and apoptosis, SIRT1 ensures the balance of adipose stores in WAT. Fat storage is in part regulated by the activity of PPAR-γ on the aP2 gene, which encodes a protein that leads to fat storage. Interestingly, when mice are starved, SIRT1 is induced in WAT to suppress PPAR-γ by docking to the negative cofactors of the nuclear receptor and, at the same time, it binds the aP2 promoter, repressing its gene expression, ultimately leading to fat mobilizing in the blood stream
[63]. These epigenetic changes lead to an increase in life expectancy, similar to what has been observed in mouse models deprived of adipogenesis (e.g., with insulin receptor deficiency) or with accelerated thermogenesis
[64].
Metabolic diseases, such as obesity or liver steatosis, are accompanied by a high incidence of cancer diseases
[65][66][67][68][69][70][65,66,67,68,69,70]. Through epigenetic activities, SIRT1 is known to play a significant role in the phenomena of carcinogenesis. However, the interplay between SIRT1 and DNA repair in cells is not completely understood yet, and, in some cases, results are even controversial
[71][72][73][74][75][76][71,72,73,74,75,76]. For example, different SIRT1-induced modifications for DNA integrity preservation have been described both in normal and in cancer cells
[8].
Most of the antitumor activity of SIRT1, however, is expressed through the containment of the metabolic syndrome. In fact, the increased life expectancy due to the epigenetic regulation of SIRT1 is due to the improvement of several metabolic and aging-related diseases, such as diabetes, NAFLD, cardiomyopathy, and neurodegenerative disorders
[16][77][16,77]. More specifically, the antagonistic relationship between SIRT1 and NF-κB seems to be protective against hepatic steatosis and the metabolic energy balance under hypothalamic control
[78]. Progression in fatty liver disease could be stopped by the AMPKa2-SIRT1-PPAR-α signaling pathway and by the SIRT1-mediated NLRP3 inflammasome suppression
[79][80][81][82][79,80,81,82]. SIRT1 also exerts a beneficial effect against the development of atherosclerosis
[14] and other diabetic complications. In this regard, it has been recently proven that tubular SIRT1 attenuates diabetic albuminuria by epigenetically suppressing claudin-1 overexpression in podocytes
[83], whereas SIRT1 activators induce increased stress resistance in diabetic cardiomyopathy through the upregulation of the kinase ERK1/2 pathway and the sarcoplasmic reticulum Ca
2+-ATPase SERCA2a
[84].
Altogether, these findings suggest that many metabolic improvements are associated with the increased expression of SIRT1. Major metabolic changes, such as glycogen and fat mobilization, and the increase in gluconeogenesis and thermogenesis, along with changes in hormone levels (reduction in insulin and increase in glucagon, adipokines, and glucocorticoids levels), occur when nutritional conditions allow for an increase in the protective effects of SIRT1. In conclusion, the availability of the cofactor NAD+, closely related to nutritional intake and varying considerably between fasting and overeating states, activates SIRT1, which also mediates the action of other epigenetic modifiers.