Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Rodrigo Santos Vicente and Version 2 by Rita Xu.
Germline pathogenic variants in the Breast Cancer Genes 1 (
BRCA1
) and 2 (
BRCA2
) are responsible for Hereditary Breast and Ovarian Cancer (HBOC) syndrome. Genetic susceptibility to breast cancer accounts for 5–10% of all cases, phenotypically presenting with characteristics such as an autosomal dominant inheritance pattern, earlier age of onset, bilateral tumours, male breast cancer, and ovarian tumours, among others.
BRCA2
pathogenic variant is usually associated with other cancers such as melanoma, prostate, and pancreatic cancers. Many rearrangements of different mutations were found in both genes, with some ethnic groups having higher frequencies of specific mutations due to founder effects.
- hereditary breast and ovarian cancer syndrome
- breast cancer
- ovarian cancer
1. Introduction
According to the World Health Organization, in 2020, there were 2.3 million women diagnosed with breast cancer (BC) and 685,000 deaths worldwide. It is considered the world’s most prevalent cancer and the most frequent cause of lost disability-adjusted life by women when compared to other cancer types [1]. Furthermore, ovarian cancer (OC) is one of the most common gynecologic cancers ranking eighth in terms of cancer incidence and mortality among women globally. Also known as a “silent killer”, OC has a lower prevalence than BC, but it is three times more lethal. Most women are diagnosed with advanced stage OC, exhibiting poor prognosis and consequently much less variability in mortality between low- and high-income countries [2]. Besides, a general population screening cannot be recommended [3].
Germline mutations in Breast Cancer Gene (BRCA)1 and 2 account for a large proportion of hereditary breast and ovarian cancer (HBOC) families [4]. The HBOC is a known example of a genetic syndrome involving BRCA mutations. It can affect men and women and usually leads to BC and/or OC before age 50. Typically, it is associated with a family history of cancer, and bilateral tumours may also occur [4]. Over time, despite technical difficulties due to the large size of both genes, screening for mutation in BRCA in different populations was performed according to the familiar cancer patterns reported. Within well-defined populations across many countries, specific frequent mutations have been identified. The founder effects generally occur in a pool of reduced genetic heterogeneity and facilitate carrier detection and genetic counselling [5]. This was the case of c.156_157insAlu BRCA2 rearrangement, firstly identified in a Portuguese resident in Belgium and now recognised as a founder mutation in Portuguese HBOC families [6].
2. Genetic and Clinical Features of Hereditary Breast and Ovarian Cancer
HBOC syndrome is defined as an autosomal dominant inherited disorder in which the risk of BC and OC is higher than normal. Globally, the hereditary predisposition cases represent 5–10% of all BC and 20–25% of OC [7]. HBOC is caused by germline mutations in BRCA 1 and 2, which are suppressor genes that encode proteins involved in DNA repair [4]. Those genes are responsible for repairing DNA double-strand breaks homologous recombination (HR) that provides accurate recombination using a sister chromatid as a template, allowing genomic stability [4]. Alterations of the BRCA1/2 genes may also occur through mechanisms other than germline mutations, for example, somatic mutations or epigenetic silencing in sporadic OC or BC [8]. Several other proteins interact and cooperate with BRCA1/2 in the DNA repair process and can also be key factors in cancer susceptibility, particularly in BC and OC, that present a defect in the HR system [8]. Since BRCA1/2 mutations can have clinical and therapeutic consequences, it has been hypothesised that these alterations can also be sensitive to DNA-damaging target agents. In the BRCA1-mutated population, the risk of BC ranges from 46–87% up to age 70, while the risk of OC is 39–63%. For the BRCA2-mutated population, the lifetime risk of developing BC or OC is 38–84% and 16–27%, respectively [4]. The clinical characteristics of BRCA-associated cancers may differ from the sporadic ones. BRCA1-related breast tumours are frequently associated with a medullary histological pattern, higher grade and more likely to be oestrogen and progesterone receptors negative and without human epidermal growth factor receptor (HER2) overexpression. BRCA2-related tumours are more heterogeneous than BRCA1. It appears to have a predominance in positive hormone receptors BC, but around 16% of triple-negative tumours (TNBC) in BRCA2 carriers were described [9]. The patients with BRCA1/2 mutations have an increased risk of developing contralateral BC, around 2%, with the higher risk corresponding to the younger age of onset [4]. Also, these mutations spectrum confers a higher risk of male BC although the association is more frequently reported with the BRCA2 variant. The cancer is usually high grade in males, with hormone receptors positive and with lymph node metastases [10][11][10,11]. Studies on BC survival suggest poor survival in individuals with a BRCA mutation, but this association has not been consistent and remains a controversial topic [12][13][14][15][12,13,14,15]. The BRCA-related OC (including fallopian tube and primary peritoneal cancers) are generally serous adenocarcinomas, as opposed to mucinous or borderline tumours. Ovarian cancer is more related to BRCA1 mutations and tends to develop at an earlier age in these women [16]. Identifying these BRCA1/2 mutations is crucial to planning the treatment and follow-up of cancer patients. In BC, international recommendations suggest a bilateral mastectomy as a primary surgical treatment in BRCA carriers because of their high rate of ipsilateral and contralateral BC. These patients’ systemic treatment options are increased, mostly due to the emerging poly (ADP-ribose) polymerase (PARP) inhibitors [17]. These drugs blocking PARP action and deficient BRCA synergistically lead to a failure in DNA repair and, consequently, to the death of tumour cells (synthetic lethality). Actually, in consequence of demonstrated benefits of PARP inhibitors, there is evidence for using these drugs in early and advanced BC settings and also in OC or fallopian tubes or primary peritoneal cancer [18][19][18,19]. Four PARP inhibitors are approved by U.S. Food and Drug Administration (FDA) and by European Medicines Agency (EMA): Olaparib, rucaparib, niraparib and talazoparib. Olaparib was the first PARP inhibitor to be approved, in 2014, as maintenance therapy for platinum-sensitive relapsed advanced OC with germline BRCA 1/2 mutations, and these results were confirmed by the SOLO-2 trial [20]. More recently, the SOLO-1 trial demonstrated the benefit of using olaparib as maintenance therapy after chemotherapy in BRCA 1/2 mutated patients [21]. Other PARP inhibitors have been approved for the maintenance treatment of recurrent, epithelial ovarian, fallopian tube, or primary peritoneal cancer irrespective of the BRCA status, such as rucaparib and niraparib. For BC, olaparib was first approved in 2018 for treating patients with germline BRCA1/2 mutations HER2-negative metastatic BC. The benefit of this approach was demonstrated in the phase III OlympiAD study that assessed olaparib monotherapy versus chemotherapy [18]. In the same setting of BC disease, talazoparib was studied in the EMBRACA trial and also demonstrated a survival benefit [19]. In the early-stage BC setting, the OlympiA trial reported a significant benefit for patients with a high risk of recurrence and germline BRCA1/2 mutations treated with adjuvant olaparib following the completion of standard therapy [22]. BRCA testing was previously used in BC only to predict the risk of future cancers and guide surgical therapies. However, with the recent advent of PARP inhibitors, it may be reasonable to consider genetic testing more widely than past criteria have considered [23]. Many studies about the efficacy of chemotherapy have reported high sensitivity in patients with BRCA mutations. Chemotherapy agents that have a mechanism of action for DNA damage, such as anthracyclines and cyclophosphamide, or DNA replication, such as platinum compounds, may explain the high effectiveness in BRCA mutated patients [7].2.1. The Founder Effect
BRCA1/2 is the most frequent alteration associated with HBOC and can affect all ethnic groups. The frequency of BRCA1/2 alteration in the general population has been estimated at one in 400–500 [4]. More than 2000 different mutations have been identified since the discovery of these genes. The most frequent mutations are small insertion/deletion frameshift, non-synonymous truncation and splice-site disruption leading to entirely non-functional BRCA proteins [24]. The incidence of mutations in high-risk families varies widely among different populations. However, in particular ethnic groups that are or were geographically or culturally isolated, specific mutations show a high frequency. This is often called the “founder effect” [4]. Founder mutations of BRCA1/2 were described in specific populations. A typical example of a founder effect is the Ashkenazi Jewish population (ancestors from Eastern and Central Europe) with a prevalence of 1:40 [25]. Founder variants in BRCA1/2 have also been reported in several additional populations, including individuals of African, Amish, and Icelandic ancestry [26]. Identifying founder mutations is informative and valuable for developing cancer gene screening panels, which help to analyse genetic susceptibility profiles [5].2.2. Portuguese Founder Mutations
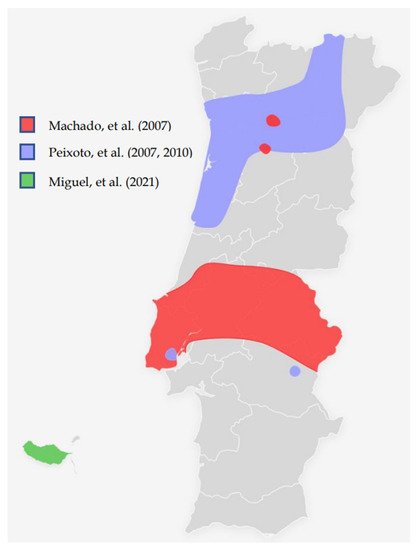
With the overseas exploration from the 15th century and after many foreign invasions occurred in the Iberia peninsula, connections with people of different cultures and genetic backgrounds were established. However, several recurrent mutations which integrate the BRCA1/2 spectrum mutations were found in Portugal. The c.156_157insAlu BRCA2 mutation is the most described in different studies (Table 1). It was first reported by Teugels, et al. in a 46-years-old Portuguese woman with BC living in Belgium with diagnosed HBOC. It was demonstrated that an Alu insertion in BRCA2 exon 3 was responsible for an in-frame deletion at the mRNA level, not detectable using the conventional mutation screening techniques [6].Table 1. Relevant studies about the prevalence of the founder mutations in Portugal.
| Studies | Aim | Number of Patients/Families | Founder Mutation Investigated | Conclusions | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Machado, et al. (2007) [27] | Molecular and phenotypic characterisation of a large insertion in exon 3 of | BRCA2. | 210 patients from Central/southern Portugal | c.156_157insAlu | c.156_157insAlu is a founder mutation of Portuguese origin and is the most frequent | BRCA2 | rearrangement | |||||
| Peixoto, et al. (2008) [28] | To evaluate the contribution of the c.156_157insAlu | BRCA2 | mutation to inherited predisposition to BC and OC in families originating mostly from northern/central Portugal. | 210 families from Northern/central Portugal |
c.156_157insAlu | This rearrangement is responsible for more than half of all pathogenic | BRCA2 | mutations and about one-fourth of all pathogenic variants in HBOC families | ||||
| Peixoto, et al. (2010) [29] | To gain insight into the ancestral origin and population spread of the c.156_157insAlu | BRCA2 | mutation. | 5443 families (149 from Portugal and 5294 from other countries than Portugal) |
c.156_157insAlu | c.156_157insAlu | BRCA2 | rearrangement is a Portuguese founder mutation that originated about 558 ± 215 years ago, accounting for the majority of the | BRCA2 | mutations and about one-third of all pathogenic germline mutations in Portuguese HBOC families. | ||
| Peixoto, et al. (2015) [30] |
To describe the mutational spectrum and evaluate the impact of founder mutations in the genetic testing criteria and strategy for molecular testing of HBOC families of Portuguese ancestry. | 1050 families (524 fully screened for | BRCA1 | / | BRCA2 | mutations) | c.156_157insAlu Other possible founder mutations pointed out: c.3331_3334del c.2037delinsCC |
Of the 119 families with pathogenic mutations, 40 (33.6%) had the | BRCA2 | c.156_157insAlu rearrangement, 15 (12.6%) the | BRCA1 | c.3331_3334del mutation and 7 (5.9%) the BRCA1 c.2037delinsCC mutation. The c.2037delinsCC mutation has not been described in other populations. |
| Miguel, et al. (2021) [31] | To evaluate the Hereditary Breast/Ovarian Cancer (HBOC) families with Madeira ancestry enrolled in the HBOC programme occurring in Instituto Português de Oncologia de Lisboa | 3566 patients (19 from Madeira Island, 3547 from other parts of Portugal) |
c.156_157insAlu | BRCA1/2 | detection rates were 27.9% and 10.5% for Madeira and the whole group, respectively. In all patients detected with BRCA1/2 mutations, 22.8 % had the c.156_157insAlu | BRCA2 | rearrangement. |