2. Helper Innate Lymphoid Cells in Melanoma
Innate lymphoid cells (ILCs) are a heterogenous group of cells characterized by three shared features: lymphoid morphology, absence of rearranged antigen-specific receptors (BCR and TCR) and lack of myeloid markers
[28]. Currently, ILCs are divided in five subsets: natural killer (NK) cells, lymphoid-tissue inducer (LTi) cells and helper ILCs, further distinguished as ILC1, ILC2 and ILC3 subsets
[20]. Based on their effector functions and expression of master transcription factors, ILCs are considered the innate counterpart of T cells
[29]. Similar to CD8+ T cells, NK cells are cytotoxic lymphocytes equipped with perforin and granzymes and able to release interferon γ (IFN-γ) and tumor necrosis factor α (TNFα) in order to kill target cells. Additionally, they require the same transcription factors, T-bet and Eomes, for their development. ILC1s, ILC2s and ILC3s parallel T helper (Th)1, Th2 and Th17 cells, respectively. ILC1s express T-bet and produce both IFN-γ and TNFα; ILC2s depend on GATA3 for their development and secrete type 2 cytokines, such as interleukin (IL)-4, IL-5 and IL-13; ILC3s recapitulate the Th17 phenotype by secreting IL-17 and IL-22 and expressing RORγt. Lastly, LTi cells, similar to ILC3s, are involved in the development of secondary lymphoid tissues during embryogenesis
[20].
As members of innate immunity, ILCs represent one of the first barriers against pathogens and malignant cells. Indeed, ILCs are able to promptly react to a wide range of stimuli, including danger signals, cytokines and stress-associated molecules expressed by tissue cells. In response, ILCs produce several different cytokines, thus playing a central role in shaping subsequent innate and adaptive immunity. Moreover, ILCs are highly plastic, which means that they can switch from a phenotype to another based on environmental signals, finely tuning immune responses
[29]. Therefore, ILCs might be important in tumorigenesis and the net effect of ILCs on the anti-tumoral immune response will depend on the recruited subpopulation, the produced cytokines and signals delivered by the TME. Of notice, all of these mechanisms might be modulated by ICIs.
2.1. ILC1s in Melanoma
Human ILC1s represent an elusive and probably heterogenous population since, except for the a-chain of the IL-7 receptor CD127, they do not express specific lineage markers and partly overlap phenotypically with NK cells
[30]. Expression levels of CD127 distinguishes ILC1s into two main subsets: CD127low ILC1s share phenotypical and functional similarities with NK cells since they respond to IL-15 and IL-12 and express prototypical NK cell markers, such as CD56, CD94 and NKp44, as well as perforin and Eomes
[31][32][31,32]; on the other hand, CD127high ILC1s lack the expression of these surface molecules and are responsive to IL-12 and IL-18
[32][33][32,33]. Therefore, ILC1s are usually considered the main helper ILC subset exerting anti-tumor activities, particularly in Th1 cytokine-enriched environments
[29].
ILC1s have been shown to be expanded in peripheral blood and infiltrated lymph nodes of melanoma patients, as well as within the tumors of melanoma-bearing mice
[24][34][24,34]. Moreover, the specific enrichment of CD56
+CD94
+ NK-like cells was observed within peripheral ILC1s
[24]. However, the molecular mechanism driving this expansion, as well as its net effect on melanoma disease, is not known. It has been demonstrated that both ILC3s and ILC2s can transdifferentiate into ILC1s based on the environmental cues. Upon IL-12 stimulation, ILC3s down-regulate the expression of RORγt and shift toward ILC1s, as indicated by IFNγ secretion and T-bet up-regulation
[32][33][35][32,33,35]. Since circulating ILC3s were concomitantly reduced in melanoma patients
[24], expanded ILC1s might be derived from the peripheral conversion of ILC3s into ILC1s driven by IL-12, of which serum levels have been found to be increased in melanoma patients
[36]. Alternatively, the altered frequencies of ILC1s and ILC3s could reflect transdifferentiation phenomena occurring within melanoma lesions due to IL-12 secretion by dendritic cells (DCs)
[32]. Similarly, IL-12 also induces an ILC2 shift towards ILC1s
[37][38][39][37,38,39]. However, alterations in peripheral ILC2 frequencies have not been reported in melanoma patients
[24][34][40][24,34,40].
An alternative, intriguing possibility raised from murine models is that ILC1s would be derived from NK cells under the effect of TGF-β, which is increased in the TME and circulation of melanoma patients
[23][41][23,41]. In these models, TGF-β signaling has been shown to promote the conversion of NK cells into NK-like ILC1s, characterized by an intermediate phenotype and high expression of inhibitory receptors
[42][43][42,43]. This conversion appeared to be dependent on the non-canonical TGF-β signaling pathway in NK cells
[43] (
Figure 1A). A similar switch of NK cells towards an ILC1 phenotype under the influence of TGF-β has also been demonstrated in a human setting. In this context, CD56bright NK cells were the subset most prone to conversion into ILC1s, and the process was further enhanced by IL-15
[44]. Although differences in total peripheral NK cell frequency have not been reported in melanoma patients, CD56bright NK cells has been found to be expanded and to correlate with a worse outcome
[22], which suggests the progressive conversion of cytotoxic CD56dim NK cells to the regulatory subset and then into ILC1s.
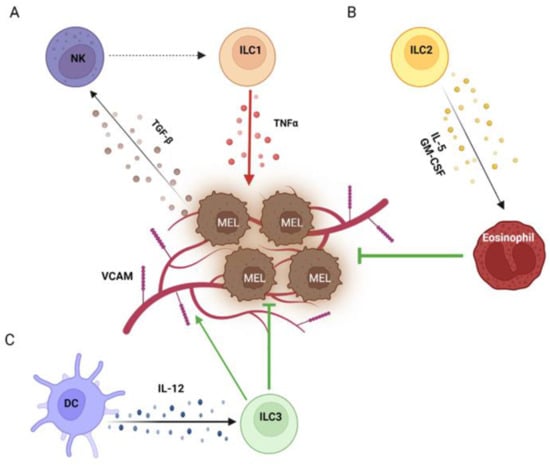
Figure 1. Helper ILCs in the melanoma TME. Within the melanoma TME, TGF-β secreted by melanoma cells switches NK cells into ILC1s, which in turn support melanoma growth by producing TNFα (A). On the other side, ILC2s exert anti-melanoma effects by recruiting eosinophils through IL-5 and GM-CSF (B). Moreover, ILC3s activated by DCs via IL-12 induce the expression of VCAM on the melanoma vasculature, promoting leukocyte infiltration (C). Created with BioRender.com
In addition, Ercolano et al. suggested that ILC1 enrichment in PBMCs from melanoma patients might be induced by melanoma cells through kynurenines, but they did not clarify whether it was due to ILC1 expansion or transdifferentiation of other subsets
[34].
In contrast with the general view of ILC1s as protective, several reports actually indicated a pro-tumoral role for this subset in melanoma. Functional data from mouse models indicated that ILC1s derived from NK cells were not only unable to counteract tumor growth and metastasization but that they even promoted it, possibly because of the increased secretion of TNF-α and pro-angiogenic factors together with the poor production of IFN-γ and CCL5, which recruit and activate T and NK cells
[42][43][42,43] (
Figure 1A). Data obtained in human settings are in line with those from murine models, showing the reduced secretion of IFN-γ and the enhanced production of TNFα by ILC1s when they were co-cultured with melanoma cells
[23][34][23,34]. The molecular mechanism by which ILC1s can interact with melanoma cells is not clear. Both murine and human ILC1s have been demonstrated to be equipped with surface receptors shared by NK cells, which recognize cognate ligands expressed by melanoma cells and affect NK cell-mediated killing
[22][23][45][22,23,45]. However, the surface receptors involved in the cross-talk between ILC1s and melanoma cells, as well as the functional effect of such interactions, are still not known.
2.2. ILC2s in Melanoma
ILC2s probably represent the best defined human ILC subset due to the specific expression of the prostaglandin D2 receptor (CRTH2)
[28]. In addition, ILC2s can express the stemness marker c-kit (CD117), which distinguishes ILC2s into two subpopulations: CD117
+ ILC2s are more plastic and share some features with ILC3s, while CD117
- ILC2s are more committed towards a Th2-associated function
[46][47][46,47]. Th2 responses have been usually associated with a tumor-supportive microenvironment due to the capability of Th2 cytokines to promote M2 polarization, the accumulation of myeloid-derived suppressor cells (MDSCs), Th2 lymphocytes and regulatory T cells (Tregs) and tissue repair. Therefore, ILC2s have been classically regarded as a pro-tumoral subset, and their abundance and/or activity have been found to be increased in several tumors
[48]. However, in the melanoma setting, experimental evidence mainly points toward an anti-tumoral role for this subset.
Although major alterations in the peripheral frequency of ILC2s in melanoma patients have not been reported
[24][34][40][24,34,40], this subset showed a low abundance within melanoma lesions in both human and murine settings
[49][50][49,50]. As previously mentioned, this could be due to the transdifferentiation of ILC2s into ILC1s and/or ILC3s following melanoma TME stimuli
[37][46][47][37,46,47]. Alternatively, ILC2 paucity could be due to TME acidification by melanoma cells, which has been shown to impair the survival and proliferation, as well as tumor infiltration, of this subset
[49].
Manipulation of ILC2s in murine melanoma models showed that this subset affected melanoma progression, which was worsened when ILC2s were depleted and improved when they were stimulated, overall suggesting that ILC2s counteract melanoma development and progression. Different mechanisms have been elucidated explaining such an effect, mainly involving the activation and recruitment of eosinophils. In 2012, Ikutani and coworkers reported that, in a mouse model of melanoma lung metastasis, ILC2s responded to melanoma cells by secreting IL-5, which in turn promoted eosinophil recruitment and activation, thus counteracting metastasization
[51] (
Figure 1B). Similar findings were later confirmed by Wagner et al., who however did not further investigate the underlying molecular mechanism
[49]. An analogous capability of ILC2s to restrain melanoma growth via eosinophil expansion and activation has also been described by Jacquelot et al. in a model of primary melanoma. However, in this context, the phenomenon was due to granulocyte–macrophage colony-stimulating factor (GM-CSF) secretion by ILC2s
[50] (
Figure 1B). In addition, two other mechanisms have been reported for ILC2s to directly restrain melanoma by inducing apoptosis thorough the secretion of the chemokines CXCL1 and CXCL2 in primary tumors and TNFα in lung metastases
[52][53][52,53], suggesting that different anti-tumoral mechanisms may be specifically induced base on the TME. In all these models, ILC2 activation was mediated by the exogenous administration of IL-33. However, it is unclear whether the intra-tumoral levels of IL-33 are actually able to activate ILC2s. Moreover, human ILC2s have been shown to be able to respond to melanoma cells in vitro by up-regulating TNFα and IL-13
[24]; thus, it is possible that cell-to-cell interactions may contribute to activation
[45].
On the other hand, a pro-tumoral role for IL-33-activated ILC2s in the context of melanoma has also been described. Particularly, two distinct murine models demonstrated that IL-33-stimulated ILC2s were able to negatively affect NK cell anti-melanoma activity and tumor rejection
[54][55][54,55]. Again, different pathways have been implied, which could occur concomitantly. Following IL-33 stimulation, ILC2s have been shown to up-regulate the ectoenzyme CD73
[54], of which the product adenosine is well known to be enriched within the melanoma TME and to suppress NK cell cytotoxicity
[29]. Moreover, ILC2s could suppress NK cells indirectly by recruiting eosinophils, which in turn limit glucose availability, needed for NK cell functions
[55].
These conflicting effects of ILC2s on melanoma observed in vivo may be due to the different protocols used to stimulate ILC2s with IL-33, largely varying in terms of the amount, way and scheduling of administration. Thus, it is possible that the same cytokine, at different concentrations, may activate distinct pathways in ILC2s with divergent effects on melanoma development and progression.
2.3. ILC3s in Melanoma
ILC3 sare identified based on the expression of the stemness marker c-kit (CD117) and the natural cytotoxicity receptors (NCRs) NKp46 (in mice) and NKp44 (in humans). Particularly, NCRs functionally distinguish ILC3s into two subsets: NCR
- ILC3s secrete IL-17, whereas the production of IL-22 is restricted to NCR
+ ILC3s
[20]. However, recent evidence suggests that ILC3s may be a heterogeneous population containing ILC precursors, as well as more mature ILC3s
[56]. Given the phenotypical and functional heterogeneity of this subset, as well as its involvement in both inflammation and tissue healing
[57], the ILC3 role in melanoma is controversial.
In the periphery of melanoma patients, ILC3s have been shown to be reduced, particularly in the NCR
+ component
[24]. As previously mentioned, the total contraction of ILC3s could be due to their conversion into ILC1s under the influence of IL-12
[32][33][35][32,33,35], while TGF-β might promote the down-regulation of NCRs
[58]. Thus, melanoma disease could trigger a complex pathway in which mature ILC3s regress into precursors under the influence of various cytokines and are then converted into ILC1s. Alternatively, ILC3s could directly switch into NK cells
[59].
In mouse models, the presence of IL-12 within the melanoma TME has been shown to promote the anti-tumor activity of NKp46
+ ILC3s against both primary tumors and metastases
[60][61][60,61] (
Figure 1C). Although NKp46
+ ILC3s acquired an ILC1/NK cell-like phenotype under the influence of IL-12, melanoma rejection was not due to a functional switch, as ILC1s were unable to control tumor growth and key molecules for ILC1 and NK cell anti-tumor activities were dispensable as well. Instead, IL-12-stimulated NKp46
+ ILC3s induced the expression of adhesion molecules, especially vascular cell adhesion molecule (VCAM), on melanoma vessels, promoting leucocyte influx
[60][61][60,61]. A similar role has also been proposed for NKp46
- ILC3s, which specifically recruited myeloid cells within melanoma
[62]. However, the underlying mechanism has not been clarified. Since (i) ILC3s have been shown in vitro to respond to melanoma cells by producing TNFα
[23], (ii) melanoma cells express NCR ligands
[63] and (iii) TNFα is able to stimulate adhesion molecule expression on endothelial cells
[64], ILC3s might likely sense melanoma cells through NCRs and also secrete TNFα in response in vivo, which in turn would promote endothelial activation and leukocyte recruitment.
However, in a CCL21-enriched TME, ILC3s have been proposed to contribute to tumor growth through the generation of a lymphoid-like stroma, which in turn would contribute to establish a tolerogenic environment
[65].