The coronavirus 2019 (COVID-19) pandemic was caused by a positive sense single-stranded RNA (ssRNA) severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Prior COVID-19 research on immunogens understandably focused on nAb responses, with less interest in overall cellular immunity. Interestingly, there is data accumulating which suggests that T cell responses are an important player in both natural and adaptive immunity as well as vaccine protection against chronic COVID-19 disease.
- SARS-CoV-2
- T-cells
- immune response
- adaptive
- innate
- covid-19
- immunology
- NK cells
1. Overview to T Cells and the Adaptive Immune System
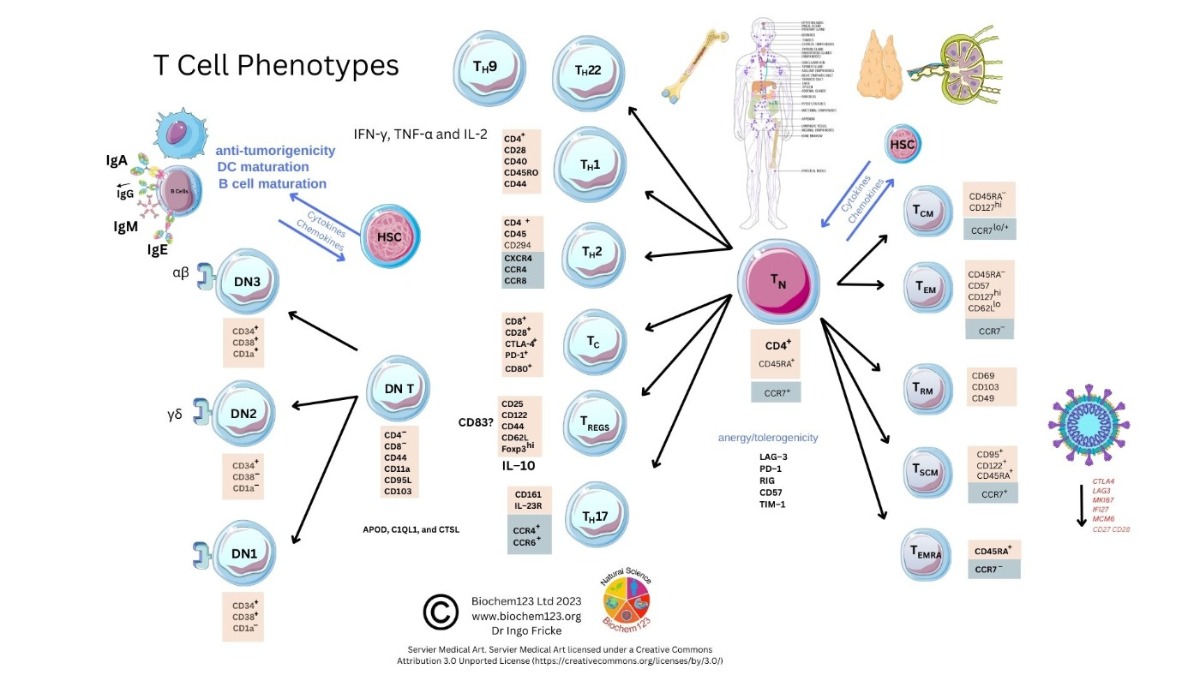
2. Background to T Cells in Coronaviruses and Host SARS-CoV-2 Infection
3. Helper T Cell Role during the Adaptive Immune Response to SARS-CoV-2
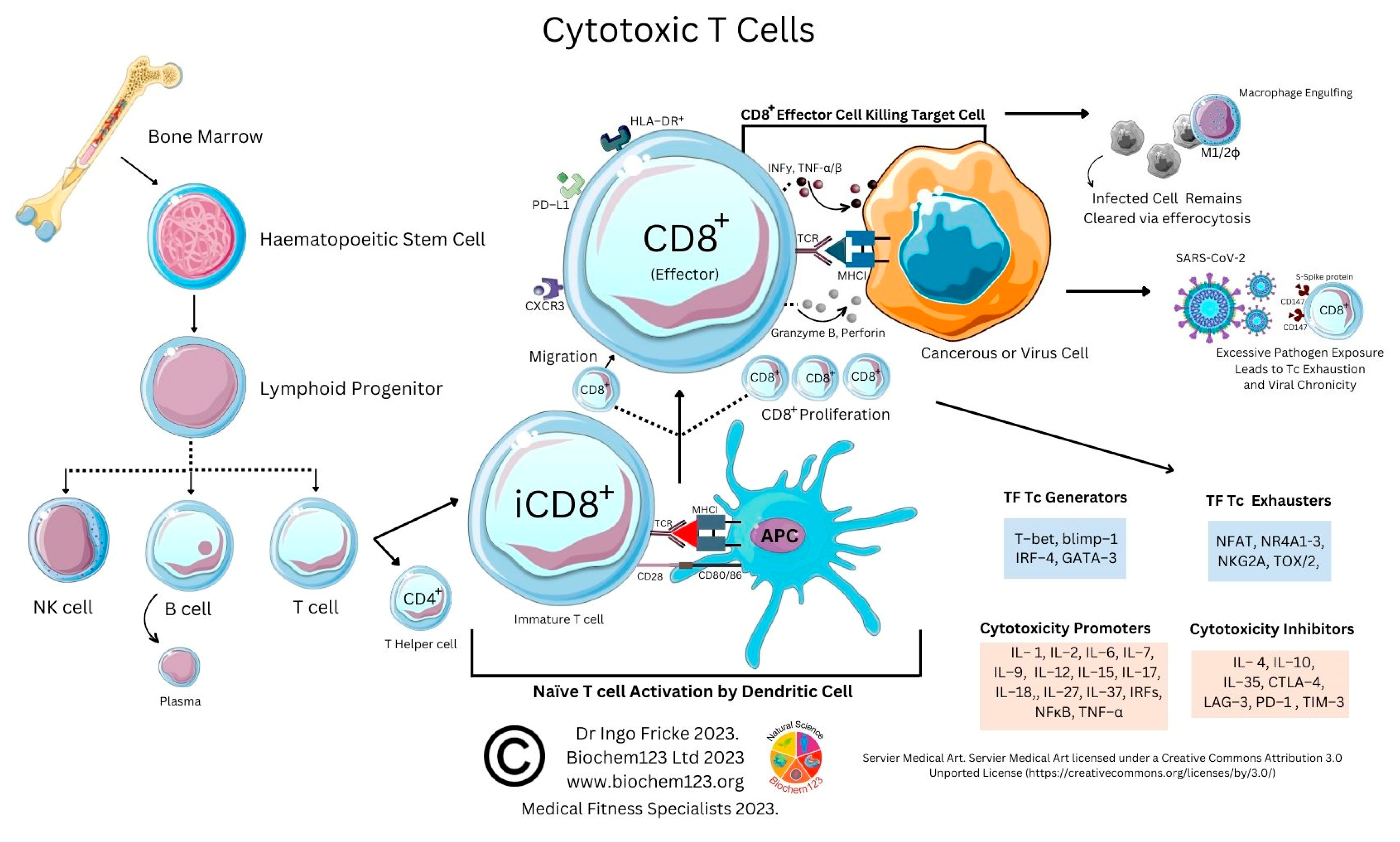
4. Cytotoxic T Cell Role during the Adaptive Immune Response to SARS-CoV-2
5. Regulatory T Cell Overview during the Adaptive Immune Response
6. TH17 Cell Overview during the Adaptive Immune Response
7. T Cell Exhaustion in COVID-19
As above, SARS-CoV-2 infection elicits the activation of both innate and adaptive immunity [90][91]. In addition to nAbs, T cell immunity plays a crucial role in virus clearance and prevention [92]. Adaptive T cells have been reported to provide some long-term immunity against COVID-19 disease [93]. Adaptive T cell immunity consists of two major cellular populations known as CD4+ and CD8+ T cells. Antigen presenting cells (APCs) possess a critical role in activation and differentiation of these T cells. In general, infected pathogens are recognized, processed, and presented by APCs. The processed immunogenic peptides are later presented by either HLA class I or HLA class II molecules expressed on APCs surface for T cell recognition via T cell receptors (TCR). Upon activation, T cells play a vital role in pathogen clearance with CD8+ cytotoxic T cells capable of secreting a wide range of molecules such as perforins, granzymes, INF-γ and apoptotic protein Fas ligand to clear pathogen-infected cells from the host [94][95]. Meanwhile, CD4+ helper T cells play an essential role in stimulation of cytokines, proliferation of CD8+ T cells and recruitment of B cells to produce nAbs to eradicate pathogens [96]. Recent evidence shows the importance of SARS-CoV-2-specific CD8+ T cell responses in protecting against development of severe COVID-19 disease in patients [94][97]. However, persistent infection by the pathogen may result in T cell exhaustion leading to functionally ineffective and/or compromised pathogen clearance [98][99][100][101][102]. T cell exhaustion due to persistent antigenic exposure has been widely reported in many chronic diseases including human immunodeficiency virus (HIV), hepatitis C virus (HCV), hepatitis B virus (HBV), cancer, and malaria [103][104][105][106][107]. Although it is still a controversial issue on the association of COVID-19 disease with immune exhaustion, some studies demonstrate a reduction in CD4+ and CD8+ T cells in ICU-admitted COVID-19 disease severe patients [97][108][109][110]. The previous evidence shows that circulating serum isolated from these severe COVID-19 patients (n = 522) had remarkably higher levels of pro-inflammatory cytokines, especially TNF-α, IL-6, and IL-10 in comparison to those with mild-moderate patients [107][109]. The uncontrollable rise of pro-inflammatory cytokines in response to COVID-19, resulting from the release of a large amount of the cytokines in the body all at once, could be a strong indicator of “cytokine storm” that causes organ failure and death in COVID-19 [111][112]. In addition, the study also indicates that severe COVID-19 disease patients possess higher ratio of inhibitory receptors (IR) expressing T cells [109]. T cell profiling demonstrates both CD4+ and CD8+ T cell subsets in severe COVID-19 disease patients that exhibit exhausted cellular phenotypes with remarkable expression of IR factors that include PD-1, NKG2A and TIM-3, suggesting exhausted T cells might not be able to perform viral clearance effectively in severe COVID-19 disease patients [108]. This could be indicative of exhausted CD8+ T cells, which may have lost their potential to differentiate into memory T cells which are important for sterilizing immunity [113][114]. In addition, characterization of cells from lung bronchoalveolar lavage fluid COVID-19 disease patients (n = 13) using single-cell RNA sequencing shows that patients with severe disease had relatively decreased levels of CD8+ T cells with an increased proportion of pro-inflammatory monocyte-derived macrophages and natural killer cells, indicating the importance of functional T cells in SARS-CoV-2 sterilizing immunity and COVID-19 recovery [114].
References
- Kokuina, E.; Breff-Fonseca, M.C.; Villegas-Valverde, C.A.; Mora-Díaz, I. Normal Values of T, B and NK Lymphocyte Subpopulations in Peripheral Blood of Healthy Cuban Adults. MEDICC Rev. 2019, 21, 16–21.
- Song, M.; Graubard, B.I.; Rabkin, C.S.; Engels, E.A. Neutrophil-to-Lymphocyte Ratio and Mortality in the United States General Population. Sci. Rep. 2021, 11, 464.
- Bernard, A.; Boumsell, L.; Hill, C. Joint Report of the First International Workshop on Human Leucocyte Differentiation Antigens by the Investigators of the Participating Laboratories. In Leucocyte Typing; Springer: Berlin/Heidelberg, Germany, 1984; pp. 9–142.
- Bulygin, A.S.; Khantakova, J.N.; Shkaruba, N.S.; Shiku, H.; Sennikov, S.S. The Role of Metabolism on Regulatory T Cell Development and Its Impact in Tumor and Transplantation Immunity. Front. Immunol. 2022, 13, 1016670.
- Jarjour, N.N.; Masopust, D.; Jameson, S.C. T Cell Memory: Understanding COVID-19. Immunity 2021, 54, 14–18.
- Wu, Z.; Zheng, Y.; Sheng, J.; Han, Y.; Yang, Y.; Pan, H.; Yao, J. CD3+CD4-CD8- (Double-Negative) T Cells in Inflammation, Immune Disorders and Cancer. Front. Immunol. 2022, 13, 816005.
- Frumento, G.; Verma, K.; Croft, W.; White, A.; Zuo, J.; Nagy, Z.; Kissane, S.; Anderson, G.; Moss, P.; Chen, F.E. Homeostatic Cytokines Drive Epigenetic Reprogramming of Activated T Cells into a “Naive-Memory” Phenotype. iScience 2020, 23, 100989.
- He, Q.; Jiang, X.; Zhou, X.; Weng, J. Targeting Cancers through TCR-Peptide/MHC Interactions. J. Hematol. Oncol. 2019, 12, 139.
- Quinti, I.; Lougaris, V.; Milito, C.; Cinetto, F.; Pecoraro, A.; Mezzaroma, I.; Mastroianni, C.M.; Turriziani, O.; Bondioni, M.P.; Filippini, M.; et al. A Possible Role for B Cells in COVID-19? Lesson from Patients with Agammaglobulinemia. J. Allergy Clin. Immunol. 2020, 146, 211–213.e4.
- Soresina, A.; Moratto, D.; Chiarini, M.; Paolillo, C.; Baresi, G.; Focà, E.; Bezzi, M.; Baronio, B.; Giacomelli, M.; Badolato, R. Two X-linked Agammaglobulinemia Patients Develop Pneumonia as COVID-19 Manifestation but Recover. Pediatr. Allergy Immunol. 2020, 31, 565–569.
- Devassikutty, F.M.; Jain, A.; Edavazhippurath, A.; Joseph, M.C.; Peedikayil, M.M.T.; Scaria, V.; Sandhya, P.; Govindaraj, G.M. X-Linked Agammaglobulinemia and COVID-19: Two Case Reports and Review of Literature. Pediatr. Allergy Immunol. Pulmonol. 2021, 34, 115–118.
- Mateus, J.; Grifoni, A.; Tarke, A.; Sidney, J.; Ramirez, S.I.; Dan, J.M.; Burger, Z.C.; Rawlings, S.A.; Smith, D.M.; Phillips, E.; et al. Selective and Cross-Reactive SARS-CoV-2 T Cell Epitopes in Unexposed Humans. Science 2020, 370, 89–94.
- Braun, J.; Loyal, L.; Frentsch, M.; Wendisch, D.; Georg, P.; Kurth, F.; Hippenstiel, S.; Dingeldey, M.; Kruse, B.; Fauchere, F.; et al. SARS-CoV-2-Reactive T Cells in Healthy Donors and Patients with COVID-19. Nature 2020, 587, 270–274.
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e15.
- Meckiff, B.J.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Kusnadi, A.; Simon, H.; Eschweiler, S.; Grifoni, A.; Pelosi, E.; Weiskopf, D.; et al. Imbalance of Regulatory and Cytotoxic SARS-CoV-2-Reactive CD4+ T Cells in COVID-19. Cell 2020, 183, 1340–1353.e16.
- Xu, Q.; Milanez-Almeida, P.; Martins, A.J.; Radtke, A.J.; Hoehn, K.B.; Oguz, C.; Chen, J.; Liu, C.; Tang, J.; Grubbs, G.; et al. Adaptive immune responses to SARS-CoV-2 persist in the pharyngeal lymphoid tissue of children. Nat. Immunol. 2023, 24, 186–199.
- Choi, S.J.; Kim, D.U.; Noh, J.Y.; Kim, S.; Park, S.H.; Jeong, H.W.; Shin, E.C. T cell epitopes in SARS-CoV-2 proteins are substantially conserved in the Omicron variant. Cell. Mol. Immunol. 2022, 19, 447–448.
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14.
- Nelde, A.; Bilich, T.; Heitmann, J.S.; Maringer, Y.; Salih, H.R.; Roerden, M.; Lübke, M.; Bauer, J.; Rieth, J.; Wacker, M.; et al. SARS-CoV-2-Derived Peptides Define Heterologous and COVID-19-Induced T Cell Recognition. Nat. Immunol. 2021, 22, 74–85.
- Bacher, P.; Rosati, E.; Esser, D.; Martini, G.R.; Saggau, C.; Schiminsky, E.; Dargvainiene, J.; Schröder, I.; Wieters, I.; Khodamoradi, Y.; et al. Low-Avidity CD4+ T Cell Responses to SARS-CoV-2 in Unexposed Individuals and Humans with Severe COVID-19. Immunity 2020, 53, 1258–1271.e5.
- Le Bert, N.; Tan, A.T.; Kunasegaran, K.; Tham, C.Y.L.; Hafezi, M.; Chia, A.; Chng, M.H.Y.; Lin, M.; Tan, N.; Linster, M.; et al. SARS-CoV-2-Specific T Cell Immunity in Cases of COVID-19 and SARS, and Uninfected Controls. Nature 2020, 584, 457–462.
- Niessl, J.; Sekine, T.; Lange, J.; Konya, V.; Forkel, M.; Maric, J.; Rao, A.; Mazzurana, L.; Kokkinou, E.; Weigel, W.; et al. Identification of Resident Memory CD8+ T Cells with Functional Specificity for SARS-CoV-2 in Unexposed Oropharyngeal Lymphoid Tissue. Sci. Immunol. 2021, 6, eabk0894.
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic Characterisation and Epidemiology of 2019 Novel Coronavirus: Implications for Virus Origins and Receptor Binding. Lancet 2020, 395, 565–574.
- Loyal, L.; Braun, J.; Henze, L.; Kruse, B.; Dingeldey, M.; Reimer, U.; Kern, F.; Schwarz, T.; Mangold, M.; Unger, C.; et al. Cross-Reactive CD4+ T Cells Enhance SARS-CoV-2 Immune Responses upon Infection and Vaccination. Science 2021, 374, eabh1823.
- Becerra-Artiles, A.; Calvo-Calle, J.M.; Co, M.D.; Nanaware, P.P.; Cruz, J.; Weaver, G.C.; Lu, L.; Forconi, C.; Finberg, R.W.; Moormann, A.M.; et al. Broadly Recognized, Cross-Reactive SARS-CoV-2 CD4 T Cell Epitopes Are Highly Conserved across Human Coronaviruses and Presented by Common HLA Alleles. Cell Rep. 2022, 39, 110952.
- Low, J.S.; Vaqueirinho, D.; Mele, F.; Foglierini, M.; Jerak, J.; Perotti, M.; Jarrossay, D.; Jovic, S.; Perez, L.; Cacciatore, R.; et al. Clonal Analysis of Immunodominance and Crossreactivity of the CD4 T Cell Response to SARS-CoV-2. Science 2021, 372, 1336–1341.
- Lu, X.; Hosono, Y.; Nagae, M.; Ishizuka, S.; Ishikawa, E.; Motooka, D.; Ozaki, Y.; Sax, N.; Maeda, Y.; Kato, Y.; et al. Identification of Conserved Sars-Cov-2 Spike Epitopes That Expand Public Ctfh Clonotypes in Mild Covid-19 Patients. J. Exp. Med. 2021, 218, e20211327.
- Bartolo, L.; Afroz, S.; Pan, Y.G.; Xu, R.; Williams, L.; Lin, C.F.; Tanes, C.; Bittinger, K.; Friedman, E.S.; Gimotty, P.A.; et al. SARS-CoV-2-Specific T Cells in Unexposed Adults Display Broad Trafficking Potential and Cross-React with Commensal Antigens. Sci. Immunol. 2022, 7, eabn3127.
- Eggenhuizen, P.J.; Ng, B.H.; Chang, J.; Fell, A.L.; Cheong, R.M.Y.; Wong, W.Y.; Gan, P.Y.; Holdsworth, S.R.; Ooi, J.D. BCG Vaccine Derived Peptides Induce SARS-CoV-2 T Cell Cross-Reactivity. Front. Immunol. 2021, 12, 3034.
- Bukhari, S.; Henick, B.S.; Winchester, R.J.; Lerrer, S.; Adam, K.; Gartshteyn, Y.; Maniar, R.; Lin, Z.; Khodadadi-Jamayran, A.; Tsirigos, A.; et al. Single-Cell RNA Sequencing Reveals Distinct T Cell Populations in Immune-Related Adverse Events of Checkpoint Inhibitors. Cell Rep. Med. 2022, 12, 100868.
- Visan, I. RORγt+ Treg Cells. Nat. Immunol. 2015, 16, 906.
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635.
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827.
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during Effector CD8 T Cell Differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 4749–4754.
- Aghbash, P.S.; Eslami, N.; Shamekh, A.; Entezari-Maleki, T.; Baghi, H.B. SARS-CoV-2 Infection: The Role of PD-1/PD-L1 and CTLA-4 Axis. Life Sci. 2021, 270, 119124.
- Maruhashi, T.; Sugiura, D.; Okazaki, I.; Okazaki, T. LAG-3: From Molecular Functions to Clinical Applications. J. Immunother. Cancer 2020, 8, e001014.
- Chocarro, L.; Blanco, E.; Zuazo, M.; Arasanz, H.; Bocanegra, A.; Fernández-Rubio, L.; Morente, P.; Fernández-Hinojal, G.; Echaide, M.; Garnica, M.; et al. Understanding LAG-3 Signaling. Int. J. Mol. Sci. 2021, 22, 5282.
- Krishnaswamy, J.K.; Gowthaman, U.; Zhang, B.; Mattsson, J.; Szeponik, L.; Liu, D.; Wu, R.; White, T.; Calabro, S.; Xu, L.; et al. Migratory CD11b + Conventional Dendritic Cells Induce T Follicular Helper Cell–Dependent Antibody Responses. Sci. Immunol. 2017, 2, eaam9169.
- André, S.; Picard, M.; Cezar, R.; Roux-Dalvai, F.; Alleaume-Butaux, A.; Soundaramourty, C.; Cruz, A.S.; Mendes-Frias, A.; Gotti, C.; Leclercq, M.; et al. T Cell Apoptosis Characterizes Severe Covid-19 Disease. Cell Death Differ. 2022, 29, 1486–1499.
- Bohan, D.; van Ert, H.; Ruggio, N.; Rogers, K.J.; Badreddine, M.; Aguilar Briseño, J.A.; Elliff, J.M.; Rojas Chavez, R.A.; Gao, B.; Stokowy, T.; et al. Phosphatidylserine Receptors Enhance SARS-CoV-2 Infection. PLoS Pathog. 2021, 17, e1009743.
- Gil-Manso, S.; Miguens Blanco, I.; López-Esteban, R.; Carbonell, D.; López-Fernández, L.A.; West, L.; Correa-Rocha, R.; Pion, M. Comprehensive Flow Cytometry Profiling of the Immune System in COVID-19 Convalescent Individuals. Front. Immunol. 2022, 12, 5734.
- Behl, T.; Kaur, I.; Aleya, L.; Sehgal, A.; Singh, S.; Sharma, N.; Bhatia, S.; Al-Harrasi, A.; Bungau, S. CD147-spike protein interaction in COVID-19: Get the ball rolling with a novel receptor and therapeutic target. Sci. Total Environ. 2022, 808, 152072.
- Sage, P.T.; Alvarez, D.; Godec, J.; von Andrian, U.H.; Sharpe, A.H. Circulating T Follicular Regulatory and Helper Cells Have Memory-like Properties. J. Clin. Investig. 2014, 124, 5191–5204.
- Hanson, A.; Cohen, H.; Wang, H.; Shekhar, N.; Shah, C.; Harvey, B.W.; Murray, R.; Harvey, C.J. Impaired ICOS Signaling between Tfh and B Cells Distinguishes Hospitalized from Ambulatory CoViD-19 patients. medRxiv, 2020; pre-print.
- Kramer, K.J.; Wilfong, E.M.; Voss, K.; Barone, S.M.; Shiakolas, A.R.; Raju, N.; Roe, C.E.; Suryadevara, N.; Walker, L.M.; Wall, S.C.; et al. Single-Cell Profiling of the Antigen-Specific Response to BNT162b2 SARS-CoV-2 RNA Vaccine. Nat. Commun. 2022, 13, 3466.
- Gazzinelli-Guimaraes, P.H.; Sanku, G.; Sette, A.; Weiskopf, D.; Schaughency, P.; Lack, J.; Nutman, T.B. Antigenic Determinants of SARS-CoV-2-Specific CD4+ T Cell Lines Reveals M Protein-Driven Dysregulation of Interferon Signaling. Front. Immunol. 2022, 13, 883159.
- Wen, C.; Dong, Z.; Wang, Y.; Ye, G.; Ma, Y.; Yi, X.; Zhou, Y.; Li, X.; Zheng, X.; Hou, J.; et al. CTLA4+CD4+CXCR5-FOXP3+ T Cells Associate with Unfavorable Outcome in Patients with Chronic HBV Infection. BMC Immunol. 2023, 24, 3.
- Asashima, H.; Mohanty, S.; Comi, M.; Ruff, W.E.; Hoehn, K.B.; Wong, P.; Klein, J.; Lucas, C.; Cohen, I.; Coffey, S.; et al. PD-1highCXCR5–CD4+ Peripheral Helper T Cells Promote CXCR3+ Plasmablasts in Human Acute Viral Infection. Cell Rep. 2023, 42, 111895.
- Hanna, S.J.; Codd, A.S.; Gea-Mallorqui, E.; Scourfield, D.O.; Richter, F.C.; Ladell, K.; Borsa, M.; Compeer, E.B.; Moon, O.R.; Galloway, S.A.; et al. T cell phenotypes in COVID-19—A living review. Oxf. Open Immunol. 2020, 2, iqaa007, PMCID:PMC7798577.
- Bar-Ephraim, Y.E.; Koning, J.J.; Burniol Ruiz, E.; Konijn, T.; Mourits, V.P.; Lakeman, K.A.; Boon, L.; Bögels, M.; van Maanen, J.P.; Den Haan, J.M.M.; et al. CD62L Is a Functional and Phenotypic Marker for Circulating Innate Lymphoid Cell Precursors. J. Immunol. 2019, 202, 171–182.
- Paulin, D.; Lilienbaum, A.; Kardjian, S.; Agbulut, O.; Li, Z. Vimentin: Regulation and Pathogenesis. Biochimie 2022, 197, 96–112.
- Arrindell, J.; Abou Atmeh, P.; Jayet, L.; Sereme, Y.; Mege, J.-L.; Desnues, B. Vimentin Is an Important ACE2 Co-Receptor for SARS-CoV-2 in Epithelial Cells. iScience 2022, 25, 105463.
- Cianciotti, B.C.; Ruggiero, E.; Campochiaro, C.; Oliveira, G.; Magnani, Z.I.; Baldini, M.; Doglio, M.; Tassara, M.; Manfredi, A.A.; Baldissera, E.; et al. CD4+ Memory Stem T Cells Recognizing Citrullinated Epitopes Are Expanded in Patients With Rheumatoid Arthritis and Sensitive to Tumor Necrosis Factor Blockade. Arthritis Rheumatol. 2020, 72, 565–575.
- Saris, A.; Reijnders, T.D.Y.; Reijm, M.; Hollander, J.C.; de Buck, K.; Schuurman, A.R.; Duitman, J.; Heunks, L.; Aman, J.; Bogaard, H.J.; et al. Enrichment of CCR6 CD8 T Cells and CCL20 in the Lungs of Mechanically Ventilated Patients with COVID-19. Eur. J. Immunol. 2021, 51, 1535–1538.
- Adam, L.; Rosenbaum, P.; Quentric, P.; Parizot, C.; Bonduelle, O.; Guillou, N.; Corneau, A.; Dorgham, K.; Miyara, M.; Luyt, C.E.; et al. CD8+PD-L1+CXCR3+ polyfunctional T cell abundances are associated with survival in critical SARS-CoV-2-infected patients. JCI Insight 2021, 6, e151571.
- Mathew, D.; Giles, J.R.; Baxter, A.E.; Oldridge, D.A.; Greenplate, A.R.; Wu, J.E.; Alanio, C.; Kuri-Cervantes, L.; Pampena, M.B.; D’Andrea, K.; et al. Deep Immune Profiling of COVID-19 Patients Reveals Distinct Immunotypes with Therapeutic Implications. Science 2020, 369, eabc8511.
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150.
- Bobcakova, A.; Barnova, M.; Vysehradsky, R.; Petriskova, J.; Kocan, I.; Diamant, Z.; Jesenak, M. Activated CD8+CD38+ Cells Are Associated With Worse Clinical Outcome in Hospitalized COVID-19 Patients. Front. Immunol. 2022, 13, 961666.
- Du, J.; Wei, L.; Li, G.; Hua, M.; Sun, Y.; Wang, D.; Han, K.; Yan, Y.; Song, C.; Song, R.; et al. Persistent High Percentage of HLA-DR+CD38high CD8+ T Cells Associated With Immune Disorder and Disease Severity of COVID-19. Front. Immunol. 2021, 12, 735125.
- Takiguchi, S.; Tomita, Y.; Uehara, S.; Tateishi, K.; Yamamoto, N.; Nakamura, M. Immunological Imprint on Peripheral Blood in Kidney Transplant Recipients after Two Doses of SARS-CoV-2 MRNA Vaccination in Japan. Front. Med. 2022, 9, 2939.
- Stephenson, E.; Reynolds, G.; Botting, R.A.; Calero-Nieto, F.J.; Morgan, M.D.; Tuong, Z.K.; Bach, K.; Sungnak, W.; Worlock, K.B.; Yoshida, M.; et al. Single-Cell Multi-Omics Analysis of the Immune Response in COVID-19. Nat. Med. 2021, 27, 904–916.
- Santopaolo, M.; Gregorova, M.; Hamilton, F.; Arnold, D.; Long, A.; Lacey, A.; Oliver, E.; Halliday, A.; Baum, H.; Hamilton, K.; et al. Prolonged T-Cell Activation and Long COVID Symptoms Independently Associate with Severe Disease at 3 Months in a UK Cohort of Hospitalized COVID-19 Patients. MedRxiv, 2022; pre-print.
- Nguyen-Contant, P.; Embong, A.K.; Kanagaiah, P.; Chaves, F.A.; Yang, H.; Branche, A.R.; Topham, D.J.; Sangster, M.Y. S Protein-Reactive IgG and Memory B Cell Production after Human SARS-CoV-2 Infection Includes Broad Reactivity to the S2 Subunit. mBio 2020, 11, e01991-20.
- Chen, Z.; Wang, Y.; Ding, X.; Zhang, M.; He, M.; Zhao, Y.; Hu, S.; Zhao, F.; Wang, J.; Xie, B.; et al. The Proportion of Peripheral Blood Tregs among the CD4+ T Cells of Autoimmune Thyroid Disease Patients: A Meta-Analysis. Endocr. J. 2020, 67, 317–326.
- Pearce, E.N.; Farwell, A.P.; Braverman, L.E. Thyroiditis. N. Engl. J. Med. 2003, 348, 2646–2655.
- Huang, S.-C.; Gau, S.-Y.; Huang, J.-Y.; Wu, W.-J.; Wei, J.C.-C. Increased Risk of Hypothyroidism in People with Asthma: Evidence from a Real-World Population-Based Study. J. Clin. Med. 2022, 11, 2776.
- Patterson, S.J.; Pesenacker, A.M.; Wang, A.Y.; Gillies, J.; Mojibian, M.; Morishita, K.; Tan, R.; Kieffer, T.J.; Verchere, C.B.; Panagiotopoulos, C.; et al. T Regulatory Cell Chemokine Production Mediates Pathogenic T Cell Attraction and Suppression. J. Clin. Investig. 2016, 126, 1039–1051.
- Wang, Y.; Zheng, J.; Islam, M.S.; Yang, Y.; Hu, Y.; Chen, X. The Role of CD4 FoxP3+ Regulatory T Cells in the Immunopathogenesis of COVID-19: Implications for Treatment. Int. J. Biol. Sci. 2021, 17, 1507–1520.
- Seepathomnarong, P.; Ongarj, J.; Sophonmanee, R.; Seeyankem, B.; Chusri, S.; Surasombatpattana, S.; Pinpathomrat, N. Regulatory T Cells Decreased during Recovery from Mild COVID-19. Viruses 2022, 14, 1688.
- Kiaee, F.; Jamaati, H.; Shahi, H.; Roofchayee, N.D.; Varahram, M.; Folkerts, G.; Garssen, J.; Adcock, I.M.; Mortaz, E. Immunophenotype and Function of Circulating Myeloid Derived Suppressor Cells in COVID-19 Patients. Sci. Rep. 2022, 12, 22570.
- Falck-Jones, S.; Österberg, B.; Smed-Sörensen, A. Respiratory and systemic monocytes, dendritic cells, and myeloid-derived suppressor cells in COVID-19: Implications for disease severity. J. Intern. Med. 2023, 293, 130–143.
- Chen, X.; Ma, W.; Zhang, T.; Wu, L.; Qi, H. Phenotypic Tfh Development Promoted by CXCR5-Controlled Re-Localization and IL-6 from Radiation-Resistant Cells. Protein. Cell 2015, 6, 825–832.
- Lee, S.H.; eun Kwon, J.; Cho, M.-L. Immunological Pathogenesis of Inflammatory Bowel Disease. Intestig. Res. 2018, 16, 26.
- Zúñiga, L.A.; Jain, R.; Haines, C.; Cua, D.J. Th17 Cell Development: From the Cradle to the Grave. Immunol. Rev. 2013, 252, 78–88.
- Karin, N. CXCR3 Ligands in Cancer and Autoimmunity, Chemoattraction of Effector T Cells, and Beyond. Front. Immunol. 2020, 11, 976.
- Yamada, A.; Arakaki, R.; Saito, M.; Tsunematsu, T.; Kudo, Y.; Ishimaru, N. Role of Regulatory T Cell in the Pathogenesis of Inflammatory Bowel Disease. World J. Gastroenterol. 2016, 22, 2195–2205.
- Oparaugo, N.C.; Ouyang, K.; Nguyen, N.P.N.; Nelson, A.M.; Agak, G.W. Human Regulatory T Cells: Understanding the Role of Tregs in Select Autoimmune Skin Diseases and Post-Transplant Nonmelanoma Skin Cancers. Int. J. Mol. Sci. 2023, 24, 1527.
- Sałkowska, A.; Karaś, K.; Karwaciak, I.; Walczak-Drzewiecka, A.; Krawczyk, M.; Sobalska-Kwapis, M.; Dastych, J.; Ratajewski, M. Identification of Novel Molecular Markers of Human Th17 Cells. Cells 2020, 9, 1611.
- Agalioti, T.; Villablanca, E.J.; Huber, S.; Gagliani, N. T H 17 cell Plasticity: The Role of Dendritic Cells and Molecular Mechanisms. J. Autoimmun. 2018, 87, 50–60.
- Honey, K. CCL3 and CCL4 actively recruit CD8+ T cells. Nat. Rev. Immunol. 2006, 6, 427.
- Baggiolini, M. CXCL8-the First Chemokine. Front. Immunol. 2015, 6, 285.
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619.
- Mishra, A.; Suman, K.H.; Nair, N.; Majeed, J.; Tripathi, V. An Updated Review on the Role of the CXCL8-CXCR1/2 Axis in the Progression and Metastasis of Breast Cancer. Mol. Biol. Rep. 2021, 48, 6551–6561.
- Reschke, R.; Gajewski, T.F. CXCL9 and CXCL10 bring the heat to tumors. Sci. Immunol. 2022, 7, eabq6509.
- Gudowska-Sawczuk, M.; Mroczko, B. What Is Currently Known about the Role of CXCL10 in SARS-CoV-2 Infection? Int. J. Mol. Sci. 2022, 23, 3673, PMCID:PMC8998241.
- Sadeghi, A.; Tahmasebi, S.; Mahmood, A.; Kuznetsova, M.; Valizadeh, H.; Taghizadieh, A.; Nazemiyeh, M.; Aghebati-Maleki, L.; Jadidi-Niaragh, F.; Abbaspour-Aghdam, S.; et al. Th17 and Treg Cells Function in SARS-CoV2 Patients Compared with Healthy Controls. J. Cell Physiol. 2021, 236, 2829–2839.
- Gurlevik, S.L.; Ozsurekci, Y.; Sağ, E.; Derin Oygar, P.; Kesici, S.; Akca, Ü.K.; Cuceoglu, M.K.; Basaran, O.; Göncü, S.; Karakaya, J.; et al. The Difference of the Inflammatory Milieu in MIS-C and Severe COVID-19. Pediatr. Res. 2022, 92, 1805–1814.
- Mahmoud Salehi Khesht, A.; Karpisheh, V.; Qubais Saeed, B.; Olegovna Zekiy, A.; Yapanto, L.M.; Nabi Afjadi, M.; Aksoun, M.; Nasr Esfahani, M.; Aghakhani, F.; Movahed, M.; et al. Different T Cell Related Immunological Profiles in COVID-19 Patients Compared to Healthy Controls. Int. Immunopharmacol. 2021, 97, 107828.
- Pérez-Martínez, A.; Martín-Quirós, A.; Ferreras, C.; Al-Akioui, K.; Mora-Rillo, M.; Goterris, R.; Briones, M.L.; Gasior, M.; Amat, P.; de Paz, R.; et al. A Phase I/II Dose-Escalation Multi-Center Study to Evaluate the Safety of Infusion of Natural Killer Cells or Memory T Cells As Adoptive Therapy in Coronavirus Pneumonia and/or Lymphopenia: (RELEASE NCT04578210). Blood 2021, 138, 1765.
- Primorac, D., et al., Adaptive Immune Responses and Immunity to SARS-CoV-2. Front Immunol, 2022. 13: p. 848582, DOI:10.3389/fimmu.2022.848582
- Jordan, S.C., Innate and adaptive immune responses to SARS-CoV-2 in humans: relevance to acquired immunity and vaccine responses. Clin Exp Immunol, 2021. 204(3): p. 310-320, DOI: 10.1111/cei.13582
- Moss, P., The T cell immune response against SARS-CoV-2. Nat Immunol, 2022. 23(2): p. 186-193, DOI: 10.1038/s41590-021-01122-w
- Wragg, K.M., et al., Establishment and recall of SARS-CoV-2 spike epitope-specific CD4(+) T cell memory. Nat Immunol, 2022. 23(5): p. 768-780, doi.org/10.1038/s41590-022-01175-5
- Rha, M.S. and E.C. Shin, Activation or exhaustion of CD8(+) T cells in patients with COVID-19. Cell Mol Immunol, 2021. 18(10): p. 2325-2333, DOI:10.1038/s41423-021-00750-4
- Ramljak, D., et al., Early Response of CD8+ T Cells in COVID-19 Patients. J Pers Med, 2021. 11(12), DOI: 10.3390/jpm11121291
- Luckheeram, R.V., et al., CD4(+)T cells: differentiation and functions. Clin Dev Immunol, 2012. 2012: p. 925135, doi.org/10.1155/2012/925135
- Kusnadi, A., et al., Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8(+) T cells. Sci Immunol, 2021. 6(55), DOI:10.1126/sciimmunol.abe4782
- Kahan SM, Wherry EJ, Zajac AJ. T cell exhaustion during persistent viral infections. Virology. 2015 May;479-480:180-93. doi: 10.1016/j.virol.2014.12.033. Epub 2015 Jan 22.
- Saeidi A, Zandi K, Cheok YY, Saeidi H, Wong WF, Lee CYQ, Cheong HC, Yong YK, Larsson M, Shankar EM. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front Immunol. 2018 Nov 9;9:2569. doi: 10.3389/fimmu.2018.02569.
- Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2009 May 26;106(21):8623-8. doi: 10.1073/pnas.0809818106. Epub 2009 May 11.
- McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol. 2019 Apr 26;37:457-495. doi: 10.1146/annurev-immunol-041015-055318. E
- Yi JS, Cox MA, Zajac AJ. T-cell exhaustion: characteristics, causes and conversion. Immunology. 2010 Apr;129(4):474-81. doi: 10.1111/j.1365-2567.2010.03255.x.
- Fenwick C, Joo V, Jacquier P, Noto A, Banga R, Perreau M, Pantaleo G. T-cell exhaustion in HIV infection. Immunol Rev. 2019 Nov;292(1):149-163. doi: 10.1111/imr.12823.
- Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, Walker B, Sullivan J, Phillips R, Pape GR, Klenerman P. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001 Jun;75(12):5550-8. doi: 10.1128/JVI.75.12.5550-5558.2001.
- Ye B, Liu X, Li X, Kong H, Tian L, Chen Y. T-cell exhaustion in chronic hepatitis B infection: current knowledge and clinical significance. Cell Death Dis. 2015 Mar 19;6(3):e1694. doi: 10.1038/cddis.2015.42. PMID: 25789969; 17. Philip M, Schietinger A. Heterogeneity and fate choice: T cell exhaustion in cancer and chronic infections. Curr Opin Immunol. 2019 Jun;58:98-103. doi: 10.1016/j.coi.2019.04.014.
- Wykes MN, Horne-Debets JM, Leow CY, Karunarathne DS. Malaria drives T cells to exhaustion. Front Microbiol. 2014 May 27;5:249. doi: 10.3389/fmicb.2014.00249.
- Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, Chen L, Li M, Liu Y, Wang G, Yuan Z, Feng Z, Zhang Y, Wu Y, Chen Y. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol. 2020 May 1;11:827. doi: 10.3389/fimmu.2020.00827.
- Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, Hippenstiel S, Dingeldey M, Kruse B, Fauchere F, Baysal E, Mangold M, Henze L, Lauster R, Mall MA, Beyer K, Röhmel J, Voigt S, Schmitz J, Miltenyi S, Demuth I, Müller MA, Hocke A, Witzenrath M, Suttorp N, Kern F, Reimer U, Wenschuh H, Drosten C, Corman VM, Giesecke-Thiel C, Sander LE, Thiel A. SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature. 2020 Nov;587(7833):270-274. doi: 10.1038/s41586-020-2598-9.
- Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, Rawlings SA, Sutherland A, Premkumar L, Jadi RS, Marrama D, de Silva AM, Frazier A, Carlin AF, Greenbaum JA, Peters B, Krammer F, Smith DM, Crotty S, Sette A. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020 Jun 25;181(7):1489-1501.e15. doi: 10.1016/j.cell.2020.05.015.
- Montazersaheb S, Hosseiniyan Khatibi SM, Hejazi MS, Tarhriz V, Farjami A, Ghasemian Sorbeni F, Farahzadi R, Ghasemnejad T. COVID-19 infection: an overview on cytokine storm and related interventions. Virol J. 2022 May 26;19(1):92. doi: 10.1186/s12985-022-01814-1.
- Mokhtari T, Hassani F, Ghaffari N, Ebrahimi B, Yarahmadi A, Hassanzadeh G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J Mol Histol. 2020 Dec;51(6):613-628. doi: 10.1007/s10735-020-09915-3.
- Wahl I, Wardemann H. Sterilizing immunity: Understanding COVID-19. Immunity. 2022 Dec 13;55(12):2231-2235. doi: 10.1016/j.immuni.2022.10.017.
- Cañete PF, Vinuesa CG. COVID-19 Makes B Cells Forget, but T Cells Remember. Cell. 2020 Oct 1;183(1):13-15. doi: 10.1016/j.cell.2020.09.013.
- Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Cheng L, Li J, Wang X, Wang F, Liu L, Amit I, Zhang S, Zhang Z. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. 2020 Jun;26(6):842-844. doi: 10.1038/s41591-020-0901-9.