Primary blast lung injury (PBLI), caused by exposure to high-intensity pressure waves from explosions in war, terrorist attacks, industrial production, and life explosions, is associated with pulmonary parenchymal tissue injury and severe ventilation insufficiency. PBLI patients, characterized by diffused intra-alveolar destruction, including hemorrhage and inflammation, might deteriorate into acute respiratory distress syndrome (ARDS) with high mortality.
- blast injuries
- acute lung injury
- hemorrhage
- inflammation
- blood coagulation
- therapeutics
1. Introduction
2. Pathophysiological Performance of PBLI
The clinical manifestations of PBLI are similar to those of pneumonia and hemorrhagic pneumonia. The specific phases and pathological manifestations are shown in Table 1.|
Phases and Timing |
Main Pathological Manifestations of PBLI |
|---|---|
|
Phase 1 [2]0–3 h |
Pulmonary hemorrhage |
|
|
|
Phase 2 [3]4–24 h |
Inflammation |
|
|
|
Phase 3 [4]After 24 h |
Hypercoagulation |
|
3. Crosstalk among Hemorrhage, Inflammation, and Coagulation
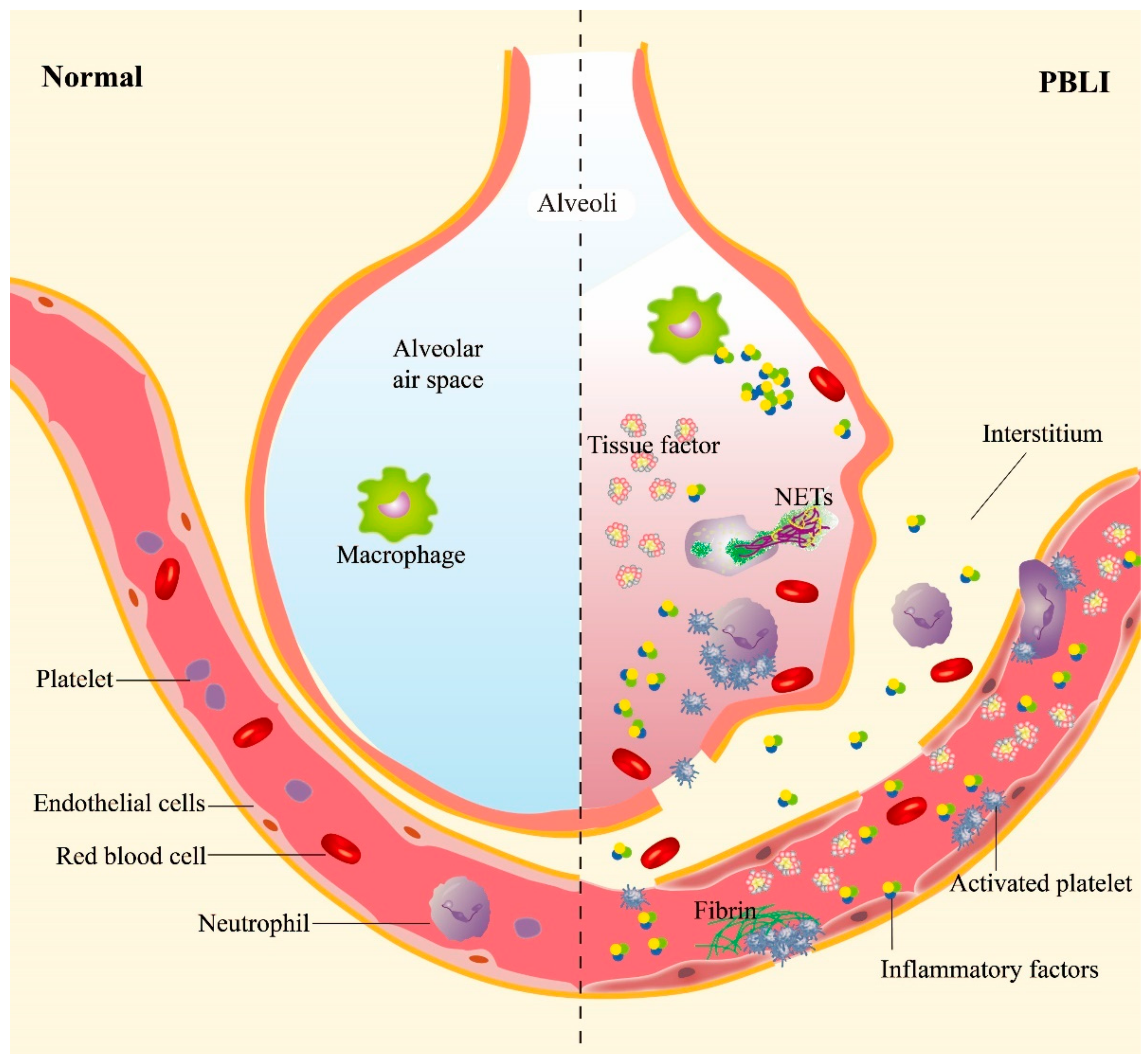
3.1. Hemorrhage Promotes Inflammation in PBLI

3.2. Inflammation Aggravates Coagulation Disorders
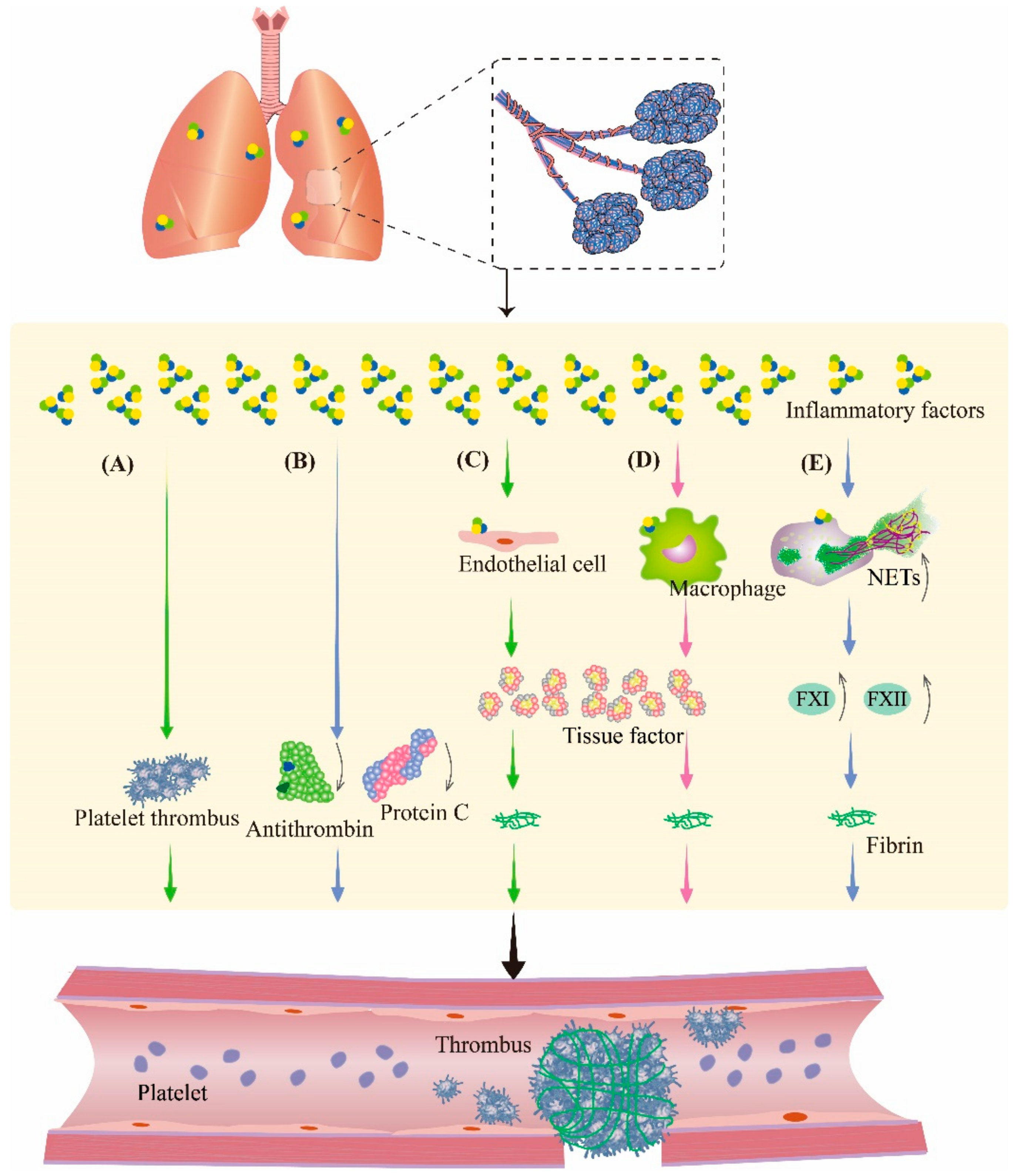
4. Pharmacotherapy Principles for PBLI
|
Drug/Efficacy |
Drug/Strategy |
Protective Mechanism |
Model |
Author |
Author Country |
Journal Year |
References |
|---|---|---|---|---|---|---|---|
|
Hemostasis |
Recombinant activated factor VII (rFVIIa) |
Coagulation factor, promotes the production of thrombin |
BLI patients |
Martinowitz et al. |
Israel |
2004 |
|
|
Tranexamic acid (TXA) |
Anti-fibrinolytic agent, impairs fibrinolysis, inhibits clot decomposition |
Adult trauma patients |
Roberts |
UK |
2015 |
||
|
Fibrinogen γ-chain-coated adenosine 5′-diphosphate-encapsulated liposomes(H12-(ADP)-liposomes) |
Targets the injured site, inhibits internal bleeding |
BLI mice |
Hagisawa et al. |
Japan |
2016 |
||
|
Thrombin@Fe3O4 nanoparticles |
Targets the damaged site, promotes the coagulation cascade |
- |
- |
- |
- |
- |
|
|
Hemostatic dexamethasone nanoparticles (hDNP) |
Targets the bleeding site, exerts anti-inflammatory effects |
BLI rats |
Hubbard et al. |
US |
2018 |
||
|
Anti-inflammation |
Sivelestat sodium hydrate (sivelestat) |
Reduces the expression of NE and IL-8 |
Severe burns rats |
Xiao et al. |
China |
2016 |
|
|
Ulinastatin |
Reduces the infiltration of inflammatory cells, reduces pulmonary edema and neutrophil infiltration, alleviates lung injury |
Rats with severe burn–blast combined injury |
Liu et al. |
China |
2018 |
||
|
BLI rabbits |
Yuan et al. |
China |
2016 |
||||
|
BLI rabbits |
Dai et al. |
China |
2015 |
||||
|
Perfluorocarbon (PFC) |
Inhibits proinflammatory cytokine release and oxidative stress |
BLI cells |
Zhang et al. |
China |
2017 |
||
|
BLI canine |
Zhang et al. |
China |
2020 |
||||
|
N-Acetylcysteine amide (NACA) |
Decreases myeloperoxidase activity, reduces NF-κB activation, attenuates lung inflammation |
PBLI rats |
Chavko et al. |
US |
2009 |
||
|
Anticoagulation |
Heparin |
Prevents diffuse intravascular coagulation, improves survival |
Trauma patients and blast injury rats |
Yang et al. |
US |
2022 |
|
|
rTFPI |
Inhibits coagulation cascade |
Gas explosion rats |
Tian et al. |
China |
2020 |
References
- Smith, J.E.; Garner, J. Pathophysiology of primary blast injury. J. R. Army Med. Corps 2019, 165, 57–62.
- Scott, T.E.; Kirkman, E.; Haque, M.; Gibb, I.E.; Mahoney, P.; Hardman, J.G. Primary blast lung injury—A review. Br. J. Anaesth. 2017, 118, 311–316.
- Peng, L.H.; Guo, G.H. Advances in the research of blast lung injury. Zhonghua Shao Shang Za Zhi 2016, 32, 156–159.
- Ritenour, A.E.; Baskin, T.W. Primary blast injury: Update on diagnosis and treatment. Crit. Care Med. 2008, 36 (Suppl. S7), S311–S317.
- Valiyaveettil, M.; Alamneh, Y.; Wang, Y.; Arun, P.; Oguntayo, S.; Wei, Y.; Long, J.B.; Nambiar, M.P. Contribution of systemic factors in the pathophysiology of repeated blast-induced neurotrauma. Neurosci. Lett. 2013, 539, 1–6.
- Zarbock, A.; Polanowska-Grabowska, R.K.; Ley, K. Platelet-neutrophil-interactions: Linking hemostasis and inflammation. Blood Rev. 2007, 21, 99–111.
- Yang, J.; Furie, B.C.; Furie, B. The biology of P-selectin glycoprotein ligand-1: Its role as a selectin counterreceptor in leukocyte-endothelial and leukocyte-platelet interaction. Thromb Haemost. 1999, 81, 1–7.
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998, 391, 591–594.
- Hermann, A.; Rauch, B.H.; Braun, M.; Schrör, K.; Weber, A.A. Platelet CD40 ligand (CD40L)—Subcellular localization, regulation of expression, and inhibition by clopidogrel. Platelets 2001, 12, 74–82.
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ. Res. 2009, 104, 346–354.
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.; Nestel, F.P.; Ni, H.; Lazarus, A.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood 2006, 107, 637–641.
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469.
- Wu, X.J.; Liu, H.M.; Song, X.M.; Zhao, B.; Leng, Y.; Wang, E.Y.; Zhan, L.; Meng, Q.; Xia, Z. Penehyclidine hydrochloride inhibits TLR4 signaling and inflammation, and attenuates blunt chest trauma and hemorrhagic shock-induced acute lung injury in rats. Mol. Med. Rep. 2018, 17, 6327–6336.
- Saha, B.K. Idiopathic pulmonary hemosiderosis: A state of the art review. Respir. Med. 2021, 176, 106234.
- Simões, R.L.; Arruda, M.A.; Canetti, C.; Serezani, C.H.; Fierro, I.M.; Barja-Fidalgo, C. Proinflammatory responses of heme in alveolar macrophages: Repercussion in lung hemorrhagic episodes. Mediat. Inflamm. 2013, 2013, 946878.
- Theofilis, P.; Sagris, M.; Antonopoulos, A.S.; Oikonomou, E.; Tsioufis, C.; Tousoulis, D. Inflammatory Mediators of Platelet Activation: Focus on Atherosclerosis and COVID-19. Int. J. Mol. Sci. 2021, 22, 11170.
- Bester, J.; Pretorius, E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci. Rep. 2016, 6, 32188.
- Van der Poll, T.; Levi, M.; Hack, C.E.; Cate, H.; van Deventer, S.J.; Eerenberg, A.J.; De Groot, E.R.; Jansen, J.; Gallati, H.; Büller, H.R. Elimination of interleukin 6 attenuates coagulation activation in experimental endotoxemia in chimpanzees. J. Exp. Med. 1994, 179, 1253–1259.
- Boermeester, M.A.; van Leeuwen, P.A.; Coyle, S.M.; Wolbink, G.J.; Hack, C.E.; Lowry, S.F. Interleukin-1 blockade attenuates mediator release and dysregulation of the hemostatic mechanism during human sepsis. Arch. Surg. 1995, 130, 739–748.
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J.; et al. Interleukin 1 receptor 1 and interleukin 1β regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arter. Thromb. Vasc. Biol. 2014, 34, 552–564.
- Eslamifar, Z.; Behzadifard, M.; Soleimani, M.; Behzadifard, S. Coagulation abnormalities in SARS-CoV-2 infection: Overexpression tissue factor. Thromb. J. 2020, 18, 38.
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arter. Thromb. Vasc. Biol. 2018, 38, 709–725.
- Osterud, B.; Rao, L.V.; Olsen, J.O. Induction of tissue factor expression in whole blood: Lack of evidence for the presence of tissue factor expression in granulocytes. Thromb. Haemost. 2000, 83, 861–867.
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885.
- Gould, T.J.; Lysov, Z.; Liaw, P.C. Extracellular DNA and histones: Double-edged swords in immunothrombosis. J. Thromb. Haemost. 2015, 13 (Suppl. S1), S82–S91.
- Meng, X.Y.; Lu, Q.Y.; Zhang, J.F.; Li, J.F.; Shi, M.Y.; Huang, S.Y.; Yu, S.F.B.; Zhao, Y.M.; Fan, H.J. A Novel Animal Model of Primary Blast Lung Injury and Its Pathological Changes in Mice. J. Trauma Acute Care Surg. 2022, 93, 530–537.
- Martinowitz, U.; Zaarur, M.; Yaron, B.L.; Blumenfeld, A.; Martonovits, G. Treating traumatic bleeding in a combat setting: Possible role of recombinant activated factor VII. Mil. Med. 2004, 169 (Suppl. S12), 16–18.
- Roberts, I. Tranexamic acid in trauma: How should we use it? J. Thromb. Haemost. 2015, 13 (Suppl. S1), S195–S199.
- Hagisawa, K.; Kinoshita, M.; Miyawaki, H.; Sato, S.; Miyazaki, H.; Takeoka, S.; Suzuki, H.; Iwaya, K.M.; Seki, S.M.; Shono, S.M.; et al. Fibrinogen γ-Chain Peptide-Coated Adenosine 5′ Diphosphate-Encapsulated Liposomes Rescue Mice From Lethal Blast Lung Injury via Adenosine Signaling. Crit. Care Med. 2016, 44, e827–e837.
- Hubbard, W.B.; Lashof-Sullivan, M.; Greenberg, S.; Norris, C.; Eck, J.; Lavik, E.; VandeVord, P. Hemostatic nanoparticles increase survival, mitigate neuropathology and alleviate anxiety in a rodent blast trauma model. Sci. Rep. 2018, 8, 10622.
- Xiao, X.G.; Zu, H.G.; Li, Q.G.; Huang, P. Sivelestat sodium hydrate attenuates acute lung injury by decreasing systemic inflammation in a rat model of severe burns. Eur. Rev. Med. Pharm. Sci. 2016, 20, 528–536.
- Liu, W.; Chai, J. Influences of ulinastatin on acute lung injury and time phase changes of coagulation parameters in rats with burn-blast combined injuries. Chin. J. Burns 2018, 34, 32–39.
- Yuan, L.; Dai, Z.; Shi, Y.; Xu, A.; Zou, Z.; Lu, Z. The effect of ulinastatin on blood gas analysis in rabbits with acute lung injury following a blast. Chin. J. Emerg. Med. 2016, 25, 301–304.
- Dai, Z.; Yuan, L.; Zou, Z.; Lu, Z.; Shi, Y. Therapeutic effect of ulinastatin combined with dexamethasone on blast-induced acute lung injury in rabbits. Chin. J. Trauma 2015, 31, 461–466.
- Zhang, Z.; Liang, Z.; Li, H.; Li, C.; Yang, Z.; Li, Y.; She, D.; Cao, L.; Wang, W.; Liu, C.; et al. Perfluorocarbon reduces cell damage from blast injury by inhibiting signal paths of NF-κB, MAPK and Bcl-2/Bax signaling pathway in A549 cells. PLoS ONE 2017, 12, e0173884.
- Zhang, Z.; Li, H.; Liang, Z.; Li, C.; Yang, Z.; Li, Y.; Cao, L.; She, Y.; Wang, W.; Liu, C.; et al. Vaporized perfluorocarbon inhalation attenuates primary blast lung injury in canines by inhibiting mitogen-activated protein kinase/nuclear factor-κB activation and inducing nuclear factor, erythroid 2 like 2 pathway. Toxicol. Lett. 2019, 319, 49–57.
- Chavko, M.; Adeeb, S.; Ahlers, S.T.; McCarron, R.M. Attenuation of pulmonary inflammation after exposure to blast overpressure by N-acetylcysteine amide. Shock 2009, 32, 325–331.
- Yang, Z.; Simovic, M.O.; Edsall, P.R.; Liu, B.; Cancio, T.S.; Batchinsky, A.I.; Cancio, L.C.; Li, Y. HMGB1 Inhibition to Ameliorate Organ Failure and Increase Survival in Trauma. Biomolecules 2022, 12, 101.
- Tian, L.Q.; Guo, Z.H.; Meng, W.Z.; Li, L.; Zhang, Y.; Yin, X.H.; Lai, F.; Li, Y.; Feng, L.; Shen, F.; et al. The abnormalities of coagulation and fibrinolysis in acute lung injury caused by gas explosion. Kaohsiung J. Med Sci. 2020, 36, 929–936.