Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Markus A. Weigand and Version 2 by Catherine Yang.
Notch signaling, a highly conserved pathway in mammals, is crucial for differentiation and homeostasis of immune cells. The spectrum of diseases is as broad as the cellular functions controlled by Notch signaling. In various types of cancer, cerebrovascular diseases, and inherited disease syndromes, Notch signaling has been found to exert a detrimental impact as well as in inflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosus (SLE), systemic sclerosis (SSc), primary biliary cirrhosis, and atherosclerosis.
- immune cells
- immune response
- Notch
- therapy
1. Notch Signaling Pathway
In mammals there are four Notch receptors (Notch1-4) and five Notch ligands [Jagged-1, Jagged-2, Delta-like 1 (DLL1), DLL3, and DLL4]. The ligands have both a membrane-bound and soluble form [1][2]. Both the receptor as well as its ligands are transmembrane proteins with abundant extracellular domains [2][12]. Ligand binding leads to a conformational change of the receptor and thus facilitates two consecutive proteolytic processes. The first cleavage is arbitrated by the A dys integrin and metalloproteases (ADAM)-family which leads to shedding of the extracellular domain. The second cleavage takes place inside the transmembrane domain and is catalyzed by a γ-secretase complex. The Notch intracellular domain (NICD) is cleaved and subsequently translocates to the nucleus to generate a transactivation complex [3][13]. This complex is composed of the deoxyribonucleic acid (DNA)-binding C-promoter-binding factor CBF-1 (CSL)/recombination signal-binding protein Jκ (RBP-J), a recombination signal sequence binding protein for Jκ genes) in mammals [4][14], and Mastermind-like protein (MAML) as a coactivator protein [5][15]. This process removes co-repressing complexes, mobilizes co-activators such as mastermind proteins, and ultimately leads to transcription of Notch target genes (Figure 1) [2][12]. The primary Notch target genes include two families of transcriptional factors hairy and enhancer of split (Hes), including HES1 and HES5, and hairy/enhancer-of-split related with YRPW motif (Hey), including HEY1 and HEY2. Other Notch target genes are B Cell lymphoma 1 Protein (BCL-1), cyclin dependent kinase inhibitor (CDKN1A), GATA binding protein 3 (GATA3), and Pre-T cell antigen receptor alpha (PTCRA) [6][5]. The interaction of Notch receptors with its ligands can be translationally modulated in the Golgi complex by O-linked glycosylation of the receptors. These post-translational modifications are initiated by the enzyme GDP-fucose Protein O-fucosyl transferase 1 (POFUT1), which adds fucose to small cystine-rich motifs called epidermal growth factor (EGF)-like repeats of the Notch extracellular domain. Additional sugar residues can be added to the fucose by glycosyltransferases, including members of the Fringe family proteins. In mammals, there are three Fringe enzymes referred to as Lunatic (Lfng), Manic (Mfng), and Radical Fringe [7][16]. These Fringe proteins provide for addition of N-acetylglucosamine residues to the glycan chain. Notch receptor glycosylation by Lfng and Mfng results in increased activation by DLL and decreased activation by Jagged ligands, while glycosylation by Radical Fringe enhances activation by all Notch ligands [8][17]. Besides this canonical Notch pathway, there are RBP-J independent non-canonical Notch signaling pathways [9][18]. These are reviewed extensively elsewhere [9][10][11][12][18,19,20,21].
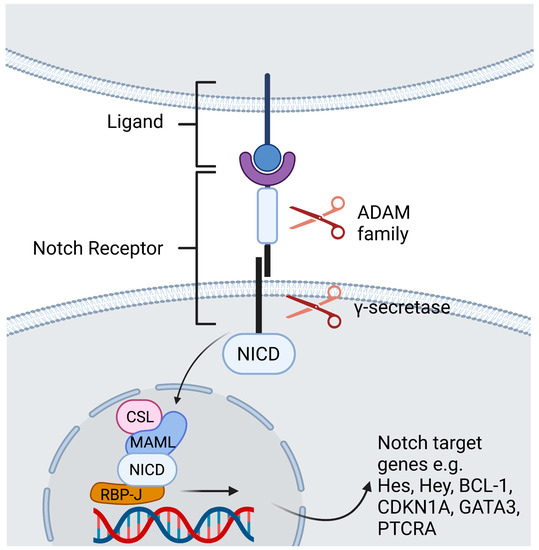
Figure 1. Contact dependent Notch signaling between cells. Ligand binding leads to a conformational change of the receptor. The first cleavage is arbitrated by the ADAM -family which leads to shedding of the extracellular domain. The second cleavage takes place inside the transmembrane domain and is catalyzed by a γ-secretase complex, that discharges the NICD to translocate to the nucleus. Within the nucleus, the NICD binds the DNA-binding protein RBP-J. Binding of NICD leads to transcription of the Notch target genes. Created with BioRender.com.
2. Notch Signaling in Leukemia and Cancer
Notch signaling as an important part of immune regulation was first described in a disease context. Aster et al. discovered that the Notch1 gene leads to T-lineage acute lymphoblastic leukemia (T-ALL) due to (7;9) chromosomal translocation of Notch1 to the TCR loci, inducing the expression of truncated forms of Notch1 [13][14][148,149]. The truncation of the Notch1 extracellular domain enables constitutive production of NICD1 in the absence of ligand binding [15][16][150,151]. Additionally, activating mutations of Notch3 have been identified by screening primary T-ALL tumors and orthotopic patient-derived xenograft models, even in the absence of activated Notch1 [17][152].
Since then, Notch signaling has been found to be associated with both pro- and anti-tumorigenic functions in various types of cancers, depending on tissue and cell type [9][18]. The role of Notch as an oncogene is well characterized for many lymphoid malignancies such as T-ALL, B-chronic lymphocytic leukemia, and splenic marginal zone lymphoma. In contrast, there is increasing evidence that Notch signaling acts as a tumor suppressor in myeloid malignancies [18][41]. However, in solid tumors such as breast cancer, lung adenocarcinoma, hepatocellular cancer, ovarian cancer, and colorectal cancer activation of Notch has been identified to be oncogenic [19][153].
3. Notch Signaling in Autoimmune Diseases
Rheumatoid arthritis is an autoimmune disease that primarily affects joints and has a prevalence of approximately 1% of the worldwide population [20][154]. The impact of Notch on arthritogenic inflammation is multifaceted but governs inflammatory events like endothelial activation, pathologic angiogenesis, as well as leukocyte recruitment, activation, and function. Notch1-initiated signaling participates in hypoxia-induced angiogenesis and conceivably also in VEGF/angiopoietin 2 (VEGF/Ang2)-induced expression of IL-6, IL-8, and Matrix metalloproteinases (MMP) 2 and 9 [21][155]. Endothelial DLL1 modulates Notch2-mediated differentiation of monocytes involved in both initiation and progression of experimental arthritis [22][156].
SLE is a systemic autoimmune disease affecting different organ systems due to a deposition of immune complexes activating the complement system [23][157]. Cleaved Notch1, cleaved Notch2, and Jagged-1 are expressed on podocytes in protein uric nephropathies including lupus nephritis, one of the most serious manifestations of SLE [24][142]. Stronger mechanistic data revealed that Notch3 affects the progression of nephritis by promoting migration and pro-inflammatory pathways [25][158]. In line with these findings, constant Notch activation results in podocyte death and it is suggested that Notch acts as a regulator of regeneration in glomerular disorders [26][159].
SSc is a chronic fibrotic disease of unknown etiology that involves the skin, and diverse internal organs [27][144]. The resulting fibrosis disturbs the physiological structure of the affected tissues, disrupts proper organ function, and is the major cause of death in SSc patients [27][28][144,160]. The Notch pathway is thought to be implicated in the fibrosis that characterizes SSc. Indeed, in the lesioned skin of SSc patients and in their fibroblasts, activated Notch1 can be found [29][30][161,162]. Mice with reactive oxygen species (ROS)-induced SSc also display elevated levels of NICD, overexpression of the ligand Jagged-1, and increased transcription of the target gene Hes-1 in the skin and lungs [29][161]. The Notch pathway is activated in SSc and inhibition of Notch signaling with the γ-secretase inhibitor DAPT exerts potent anti-fibrotic effects in this preclinical model.
4. Notch Signaling in Chronic Inflammation
There are many reports of Notch dysregulation in clinical samples from patients with different chronic inflammatory diseases. In colonic mucosal biopsies from patients with ulcerative colitis, transcription levels of Notch1 and Hes1 were significantly elevated [31][163]. A gene expression analysis revealed that Jagged-1 is expressed on endothelial cells from patients with giant cell arteritis, but not on endothelial cells from healthy individuals. Moreover, Notch1 was up regulated on circulating CD4+ T cells in these patients [32][164]. Likewise, in patients with asthma, circulating CD4+ T cells have been found to have higher Notch1 and Notch2 expression levels [33][165]. Higher levels of active Notch1 have also been observed in human appendix inflammation endothelial cells [34][166].
Atherosclerosis is an inflammatory disease characterized by the passive accumulation of lipids within artery walls [35][167]. Modified low-density lipoproteins (LDL), chronic infection, free radicals, or other factors cause a chronic inflammatory process involving the arterial endothelium [36][168]. Recently it has been shown that Notch signaling is activated in human aortal luminal endothelial cells at atherosclerotic lesions and modulates atherosclerosis by controlling macrophage polarization [37][169] into a proinflammatory phenotype [38][170]. Binesh et al. demonstrated in a rat model that enzymatic inhibition of NICD translocation by Diosgenin (a phyto steroid sapogenin) and γ-secretase inhibitor DAPT in differentiating macrophages leads to a significantly decreased NICD expression while at the same time the macrophage marker MAC387 is downregulated [39][171]. In vitro vascular inflammation models revealed that in different endothelial cells TNF promotes apoptosis through a downregulation of Notch activity. Additionally, it results in a phenotypic switch where Notch4 is replaced by Notch2. Further, Quillard et al. proved a relationship of Notch signaling, caspase activation, and apoptosis in a rat vascular inflammation model [40][172].