Mitochondria are key players in energy production, critical activity for the smooth functioning of energy-demanding organs such as the muscles, brain, and heart. Therefore, dysregulation or alterations in mitochondrial bioenergetics primarily perturb these organs. Within the cell, mitochondria are the major site of reactive oxygen species (ROS) production through the activity of different enzymes since it is one of the organelles with the major availability of oxygen.
Mitochondria are key players in energy production, critical activity for the smooth functioning of energy-demanding organs such as the muscles, brain, and heart. Therefore, dysregulation or alterations in mitochondrial bioenergetics primarily perturb these organs. Within the cell, mitochondria are the major site of reactive oxygen species (ROS) production through the activity of different enzymes since it is one of the organelles with the major availability of oxygen.
- calcium
- ROS
- mitochondria
1. Introduction
2. ROS
The Alpha and Omega of Mitochondrial ROS
3. Calcium
3.1. Influx and Efflux of Ca2+ in Mitochondria
3.2. Key Ca2+ Targets and Roles in the Regulation of Mitochondrial Bioenergetics
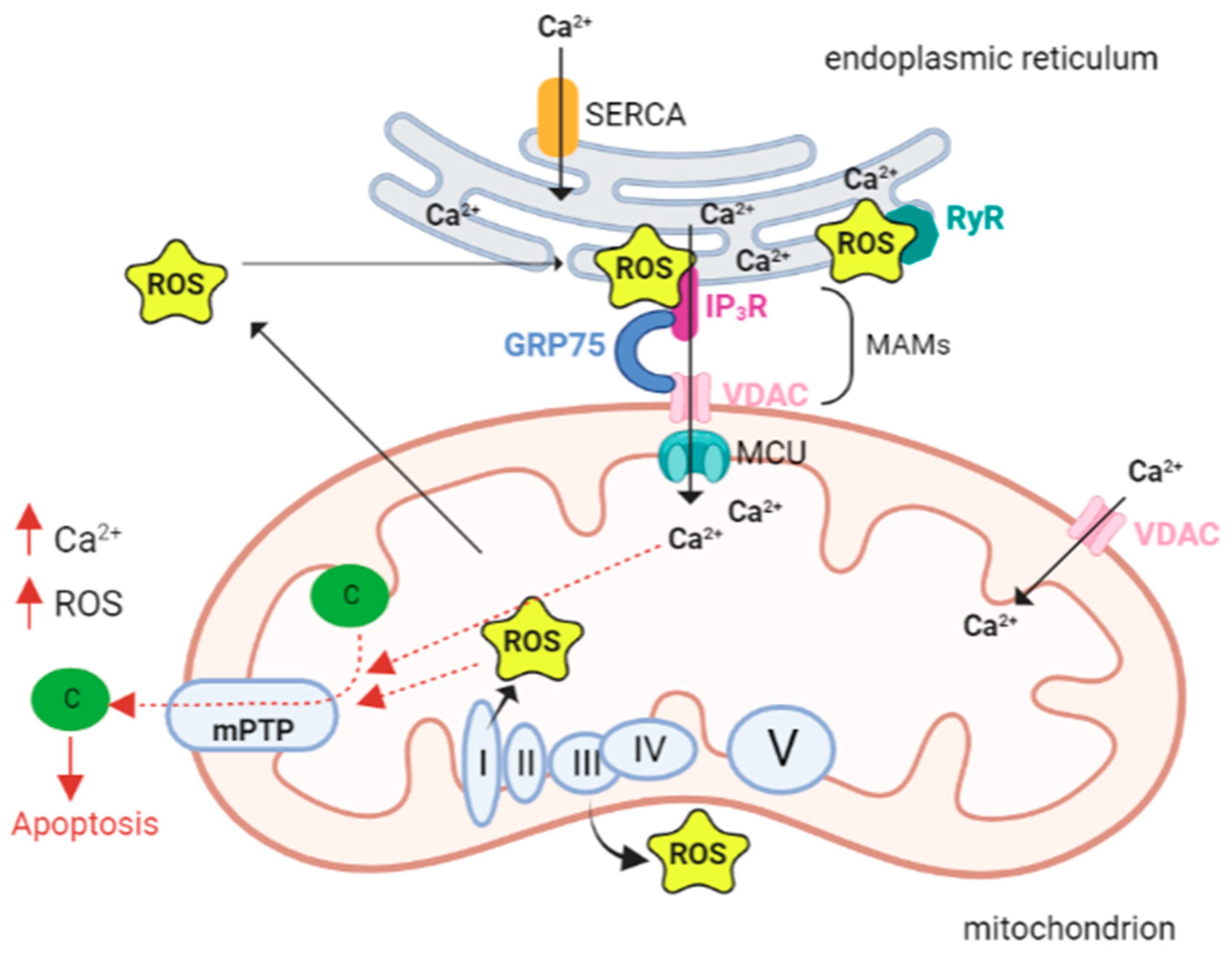
4. The Interplay between Ca2+ and ROS
References
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. In Comprehensive Physiology; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 623–634. ISBN 978-0-470-65071-4.
- Kohlhaas, M.; Nickel, A.G.; Maack, C. Mitochondrial Energetics and Calcium Coupling in the Heart. J. Physiol. 2017, 595, 3753–3763.
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13.
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The Versatility and Universality of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21.
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271.
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining Roles of Specific Reactive Oxygen Species (ROS) in Cell Biology and Physiology. Nat. Rev. Mol. Cell Biol. 2022, 23, 499–515.
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702.
- Hempel, N.; Trebak, M. Crosstalk between Calcium and Reactive Oxygen Species Signaling in Cancer. Cell Calcium 2017, 63, 70–96.
- Juan, C.A.; Pérez de la Lastra, J.M.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH Oxidases (NOX): An Overview from Discovery, Molecular Mechanisms to Physiology and Pathology. Antioxid. Basel Switz. 2021, 10, 890.
- Kuznetsov, A.V.; Margreiter, R.; Ausserlechner, M.J.; Hagenbuchner, J. The Complex Interplay between Mitochondria, ROS and Entire Cellular Metabolism. Antioxidants 2022, 11, 1995.
- Cheung, E.C.; Vousden, K.H. The Role of ROS in Tumour Development and Progression. Nat. Rev. Cancer 2022, 22, 280–297.
- Sies, H.; Chance, B. The Steady State Level of Catalase Compound I in Isolated Hemoglobin-Free Perfused Rat Liver. FEBS Lett. 1970, 11, 172–176.
- Pratt, D.A.; Tallman, K.A.; Porter, N.A. Free Radical Oxidation of Polyunsaturated Lipids: New Mechanistic Insights and the Development of Peroxyl Radical Clocks. Acc. Chem. Res. 2011, 44, 458–467.
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788.
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746.
- Paulsen, C.E.; Carroll, K.S. Cysteine-Mediated Redox Signaling: Chemistry, Biology, and Tools for Discovery. Chem. Rev. 2013, 113, 4633–4679.
- Bak, D.W.; Weerapana, E. Cysteine-Mediated Redox Signalling in the Mitochondria. Mol. Biosyst. 2015, 11, 678–697.
- Meng, J.; Fu, L.; Liu, K.; Tian, C.; Wu, Z.; Jung, Y.; Ferreira, R.B.; Carroll, K.S.; Blackwell, T.K.; Yang, J. Global Profiling of Distinct Cysteine Redox Forms Reveals Wide-Ranging Redox Regulation in C. Elegans. Nat. Commun. 2021, 12, 1415.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735.
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388.
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of Reactive Oxygen Species Generation by Mitochondria Oxidizing Different Substrates. Redox Biol. 2013, 1, 304–312.
- Fato, R.; Bergamini, C.; Leoni, S.; Lenaz, G. Mitochondrial Production of Reactive Oxygen Species: Role of Complex I and Quinone Analogues. BioFactors 2008, 32, 31–39.
- Schultz, B.E.; Chan, S.I. Structures and Proton-Pumping Strategies of Mitochondrial Respiratory Enzymes. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 23–65.
- Grgic, L.; Zwicker, K.; Kashani-Poor, N.; Kerscher, S.; Brandt, U. Functional Significance of Conserved Histidines and Arginines in the 49-KDa Subunit of Mitochondrial Complex I. J. Biol. Chem. 2004, 279, 21193–21199.
- Brand, M.D. Mitochondrial Generation of Superoxide and Hydrogen Peroxide as the Source of Mitochondrial Redox Signaling. Free Radic. Biol. Med. 2016, 100, 14–31.
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Kudin, A.P.; Bimpong-Buta, N.Y.-B.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of Superoxide-Producing Sites in Isolated Brain Mitochondria. J. Biol. Chem. 2004, 279, 4127–4135.
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial Peroxiredoxin Involvement in Antioxidant Defence and Redox Signalling. Biochem. J. 2009, 425, 313–325.
- Molavian, H.; Madani Tonekaboni, A.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and Its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, 13620.
- Angelova, P.R.; Abramov, A.Y. Functional Role of Mitochondrial Reactive Oxygen Species in Physiology. Free Radic. Biol. Med. 2016, 100, 81–85.
- Starkov, A.A.; Fiskum, G.; Chinopoulos, C.; Lorenzo, B.J.; Browne, S.E.; Patel, M.S.; Beal, M.F. Mitochondrial α-Ketoglutarate Dehydrogenase Complex Generates Reactive Oxygen Species. J. Neurosci. 2004, 24, 7779–7788.
- Finkel, T. Signal Transduction by Reactive Oxygen Species. J. Cell Biol. 2011, 194, 7–15.
- Suski, J.M.; Lebiedzinska, M.; Bonora, M.; Pinton, P.; Duszynski, J.; Wieckowski, M.R. Relation between Mitochondrial Membrane Potential and ROS Formation. Methods Mol. Biol. 2012, 810, 183–205.
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344.
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High Protonic Potential Actuates a Mechanism of Production of Reactive Oxygen Species in Mitochondria. FEBS Lett. 1997, 416, 15–18.
- Brandt, U. Energy Converting NADH:Quinone Oxidoreductase (Complex I). Annu. Rev. Biochem. 2006, 75, 69–92.
- Kussmaul, L.; Hirst, J. The Mechanism of Superoxide Production by NADH:Ubiquinone Oxidoreductase (Complex I) from Bovine Heart Mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612.
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential Effects of Mitochondrial Complex I Inhibitors on Production of Reactive Oxygen Species. Biochim. Biophys. Acta 2009, 1787, 384–392.
- Diquigiovanni, C.; Bergamini, C.; Diaz, R.; Liparulo, I.; Bianco, F.; Masin, L.; Baldassarro, V.A.; Rizzardi, N.; Tranchina, A.; Buscherini, F.; et al. A Novel Mutation in SPART Gene Causes a Severe Neurodevelopmental Delay Due to Mitochondrial Dysfunction with Complex I Impairments and Altered Pyruvate Metabolism. FASEB J. 2019, 33, 11284–11302.
- Walker, F.O. Huntington’s Disease. Semin. Neurol. 2007, 27, 143–150.
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. α-Synuclein Binds to the ER–Mitochondria Tethering Protein VAPB to Disrupt Ca2+ Homeostasis and Mitochondrial ATP Production. Acta Neuropathol. 2017, 134, 129–149.
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049.
- Patergnani, S.; Suski, J.M.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. Calcium Signaling around Mitochondria Associated Membranes (MAMs). Cell Commun. Signal. 2011, 9, 19.
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766.
- Cheng, H.; Gang, X.; He, G.; Liu, Y.; Wang, Y.; Zhao, X.; Wang, G. The Molecular Mechanisms Underlying Mitochondria-Associated Endoplasmic Reticulum Membrane-Induced Insulin Resistance. Front. Endocrinol. 2020, 11, 592129.
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative Genomics Identifies MCU as an Essential Component of the Mitochondrial Calcium Uniporter. Nature 2011, 476, 341–345.
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192.
- Yoo, J. Structural Basis of Ca2+ Uptake by Mitochondrial Calcium Uniporter in Mitochondria: A Brief Review. BMB Rep. 2022, 55, 528–534.
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of Intact Human MCU Supercomplex with the Auxiliary MICU Subunits. Protein Cell 2021, 12, 220–229.
- Marchi, S.; Pinton, P. The Mitochondrial Calcium Uniporter Complex: Molecular Components, Structure and Physiopathological Implications. J. Physiol. 2014, 592, 829–839.
- Phillips, C.B.; Tsai, C.-W.; Tsai, M.-F. The Conserved Aspartate Ring of MCU Mediates MICU1 Binding and Regulation in the Mitochondrial Calcium Uniporter Complex. eLife 2019, 8, e41112.
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.-F.; Feng, L. Structure and Mechanism of the Mitochondrial Ca2+ Uniporter Holocomplex. Nature 2020, 582, 129–133.
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial Calcium Uptake in Organ Physiology: From Molecular Mechanism to Animal Models. Pflüg. Arch.—Eur. J. Physiol. 2018, 470, 1165–1179.
- Lasorsa, F.M.; Pinton, P.; Palmieri, L.; Fiermonte, G.; Rizzuto, R.; Palmieri, F. Recombinant Expression of the Ca(2+)-Sensitive Aspartate/Glutamate Carrier Increases Mitochondrial ATP Production in Agonist-Stimulated Chinese Hamster Ovary Cells. J. Biol. Chem. 2003, 278, 38686–38692.
- Nesci, S. Mitochondrial Permeability Transition, F1 FO-ATPase and Calcium: An Enigmatic Triangle. EMBO Rep. 2017, 18, 1265–1267.
- McCormack, J.G.; Halestrap, A.P.; Denton, R.M. Role of Calcium Ions in Regulation of Mammalian Intramitochondrial Metabolism. Physiol. Rev. 1990, 70, 391–425.
- Denton, R.M. Regulation of Mitochondrial Dehydrogenases by Calcium Ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316.
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by Calcium Ions of Pyruvate Dehydrogenase Phosphate Phosphatase. Biochem. J. 1972, 128, 161–163.
- Rutter, G.A.; Denton, R.M. Regulation of NAD+-Linked Isocitrate Dehydrogenase and 2-Oxoglutarate Dehydrogenase by Ca2+ Ions within Toluene-Permeabilized Rat Heart Mitochondria. Interactions with Regulation by Adenine Nucleotides and NADH/NAD+ Ratios. Biochem. J. 1988, 252, 181–189.
- Foskett, J.K.; Mak, D.-O.D. Regulation of IP3R Channel Gating by Ca2+ and Ca2+ Binding Proteins. Curr. Top. Membr. 2010, 66, 235–272.
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283.
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P.; et al. TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Curr. Biol. CB 2018, 28, 369–382.e6.
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, Mitochondria and Cell Metabolism: A Functional Triangle in Bioenergetics. Biochim. Biophys. Acta BBA—Mol. Cell Res. 2019, 1866, 1068–1078.
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. MCUR1 Is an Essential Component of Mitochondrial Ca2+ Uptake That Regulates Cellular Metabolism. Nat. Cell Biol. 2012, 14, 1336–1343.
- Di Lisa, F.; Bernardi, P. A CaPful of Mechanisms Regulating the Mitochondrial Permeability Transition. J. Mol. Cell. Cardiol. 2009, 46, 775–780.
- Madesh, M.; Hajnóczky, G. VDAC-Dependent Permeabilization of the Outer Mitochondrial Membrane by Superoxide Induces Rapid and Massive Cytochrome c Release. J. Cell Biol. 2001, 155, 1003–1016.
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Lisa, F.D. The Mitochondrial Permeability Transition, Release of Cytochrome c and Cell Death: Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034.
- Di Lisa, F.; Bernardi, P. Mitochondrial Function and Myocardial Aging. A Critical Analysis of the Role of Permeability Transition. Cardiovasc. Res. 2005, 66, 222–232.
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950.
- Paradies, G.; Petrosillo, G.; Pistolese, M.; Di Venosa, N.; Federici, A.; Ruggiero, F.M. Decrease in Mitochondrial Complex I Activity in Ischemic/Reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ. Res. 2004, 94, 53–59.
- Nomura, K.; Imai, H.; Koumura, T.; Kobayashi, T.; Nakagawa, Y. Mitochondrial Phospholipid Hydroperoxide Glutathione Peroxidase Inhibits the Release of Cytochrome c from Mitochondria by Suppressing the Peroxidation of Cardiolipin in Hypoglycaemia-Induced Apoptosis. Biochem. J. 2000, 351, 183–193.
- Shigenaga, M.K.; Hagen, T.M.; Ames, B.N. Oxidative Damage and Mitochondrial Decay in Aging. Proc. Natl. Acad. Sci. USA 1994, 91, 10771–10778.
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria Control Store-Operated Ca2+ Entry through Na+ and Redox Signals. EMBO J. 2017, 36, 797–815.
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-Dependent Anion Channels Control the Release of the Superoxide Anion from Mitochondria to Cytosol. J. Biol. Chem. 2003, 278, 5557–5563.