During the last decades, the search for new methodologies for the synthesis of organosilicon compounds have increased due to the multiple applications in industry, in academy, among other areas. Silicon-hydrogen and silicon-silicon bond activation by d-block metals still represent the most important pathways for obtaining molecular entities that contain, witihin their structures, silane motifs. Although the chemistry of Si-H bond activation is under continue development, Si-Si bonds are poorly characterized and less used and thus, disilanes represent good starting precursors because of their stability towards ambient conditions. The present emintryi-review will focus on the physicochemical properties of Si-H and Si-Si bonds, some trends and finally highlighting their impact on the synthesis of metal complexes.
- organosilicon
- bond activation
- disilane
- hydrosilane
- oxidative addition
1. Introduction
The chemistry of metal compounds with silicon-containing ligands have been increasing over the years. The importance of the silicon atom to be incorporated as part of the resulting complexes is mainly due to the excellent σ-donor ability combined with its considerably high trans-influence/effect [1]. These features allow the metal center to be electron-rich, enabling them to activate inert substrates. Silicon (Si), placed at the the third period of the periodic table, possesses properties between carbon and their heavier analogues (Ge, Sn and Pb). The Si atom, with an electronegativity value of χSi = 1.9 in the Pauling scale, it is more electropositive than carbon (χC = 2.5), hydrogen (χH = 2.1) and many transition metals. For many bonds which incorporate silicon, their energy dissociation values show that Si−O, Si−F and Si−Cl are higher in energy, while Si−H, Si−C, Si−Si and Si−I are among the lowest in energy, as shown in Table 1.
Table 1
. Bond dissociation energy (BDE) and bond distance values for some representative silicon-containing compounds. Values obtained from reference
[2]
.
| Bond | Compound | Bond Dissociation Energy (BDE, kJ/mol) | Compound | Bond distance (pm) |
| Si−H | (CH3)3Si−H | 343 | D3Si−H | 148 |
| Si−C | (CH3)3Si−CH3 | 286 | (CH3)3Si−CH3 | 189 |
| Si−Si | (CH3)3Si−Si(CH3)3 | 300 | H3Si−SiH2F | 233 |
| ((CH3)3Si)4−Si | 236 | |||
| Si−O | (CH3)3Si−OCH3 | 530 | H3Si−O−SiH3 | 163.1 |
| (CH3)3Si−O−Si(CH3)3 | 445 | (CH3)3Si−O−Si(CH3)3 | 162.6 | |
| (CH3)3Si−OH | 430 | |||
| Si−F | (CH3)3Si−F | 590 | H3Si−F | 159.5 |
| F3Si−F | 154 | |||
| Si−Cl | (CH3)3Si−Cl | 426 | H3Si−Cl | 204.8 |
| Cl3Si−Cl | 201 | |||
| Si−Br | (CH3)3Si−Br | 334 | H3Si−Br | 220.9 |
| Br3Si−Br | 215 | |||
| Si−I | (CH3)3Si−I | 240 | H3Si−I | 243 |
| I3Si−I | 243 |
2. Si−Si Bonds Properties
Si−Si bonds properties
Si−Si bonds are non-polarized sigma bonds which possesses a mean BDE value of ca. 300 kJ/mol (72 kcal/mol), around 43 kJ/mol less than a Si−H bond. These type of bonds can be measured by spectroscopic techniques such as Nuclear Magnetic Resonance (NMR), for which Si signals tend to appear between 0 to -100 ppm, with coupling constants from 186 to 23 Hz [3]. Furthermore, Si−Si bonds are characterized for having high-energy σ and low-energy σ* molecular orbitals, which enables them to interact with electron-rich transition metals [4]. Bond distance values for Si−Si bonds are found between 235.8 and 237.2 pm [5], around 80 pm longer than in single C−C bonds, which favors the interaction between molecular orbitals of disilanes with d-atomic orbitals of metal atoms. Figure 1 shows the steric effect of substituents on some disilane compounds, with elongation or compression of the Si−Si bonds. It is observed that compounds like hexakis(tert-butyl)disilane [6] have exceptionally elongated Si−Si bonds, while compounds like H3Si−SiH3 or (CH3)3Si−Si(CH3)3 are considerably short.
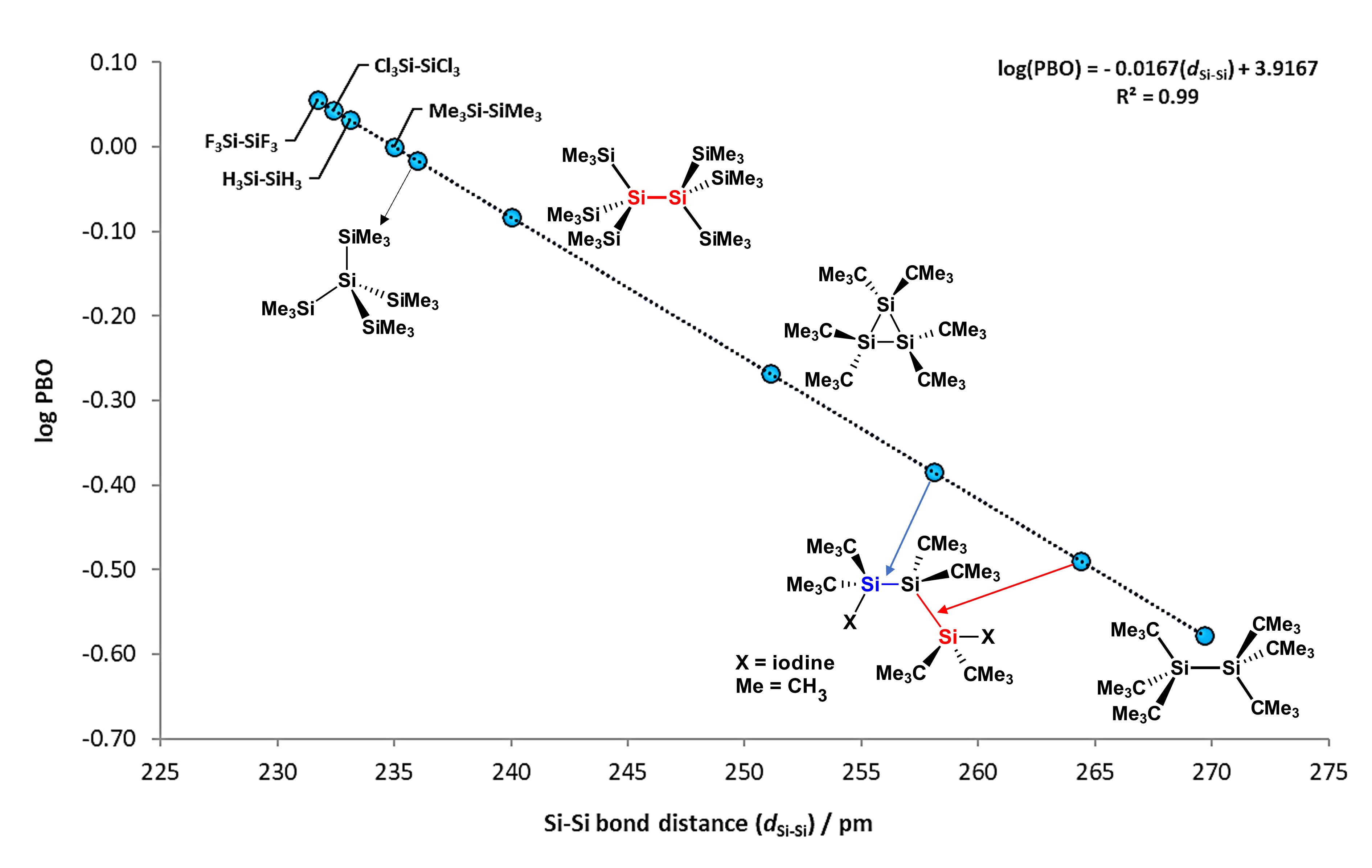
Figure 1.
Scatterplot of bond order (PBO) vs. Si−Si bond distance, where calculated log(PBO) values from experimental data is obtained from log(PBO) = (d1− d
x
)/60, where d1 = 235 pm (from Me
3
Si−SiMe
3
bond distance value set as reference) and d
x
is the Si−Si bond from selected compounds. Adapted from reference
[6].
.
Additionally, plotting the values of their distances against their Pauling "bond order" (PBO) values, mathematical equation can be obtained as follows:
log(PBO) = −0.0167(dSi−Si) + 3.9167; with R2 = 0.99
This simple relation can be used to calculate the bond order of the Si−Si bond in different disilanes, for which most of them lies between 232 and 237 pm.
The Si−Si bond is moderately reactive towards Group 1 metals (Li, Na, K, Rb, Cs). Gilman and Lichtenwalter were the first to report the cleavage of symmetrical Si−Si bonds using Li/THF [7]. Later addition of halosilanes generates unsymmetrical disilanes in moderate yields using this methodology [8]. The easiness for which Si-Si bond cleavage takes place, generally decreases as alkyl substituents attached to the Si atom increases. In general, alkyldilsilanes are not reactive towards group 1 metals. On the other hand, aryldisilanes and derivatives, (disilanes that have at least one aryl group on Si), tend to be much more reactive, as reported by Tamao and Kawachi [9]. Si-Si dissociation in these systems is considered to proceed via electronic transfer (ET) of the metal center to the low-energy σ* molecular orbitals of the disilane bond, which results in an elongation of the bond. The π-contribution between silicon and aryl groups strengthens the Si-C bond as seen in Figure 2, thus promoting specific Si-Si bond cleavage where metallic Li is used [10].
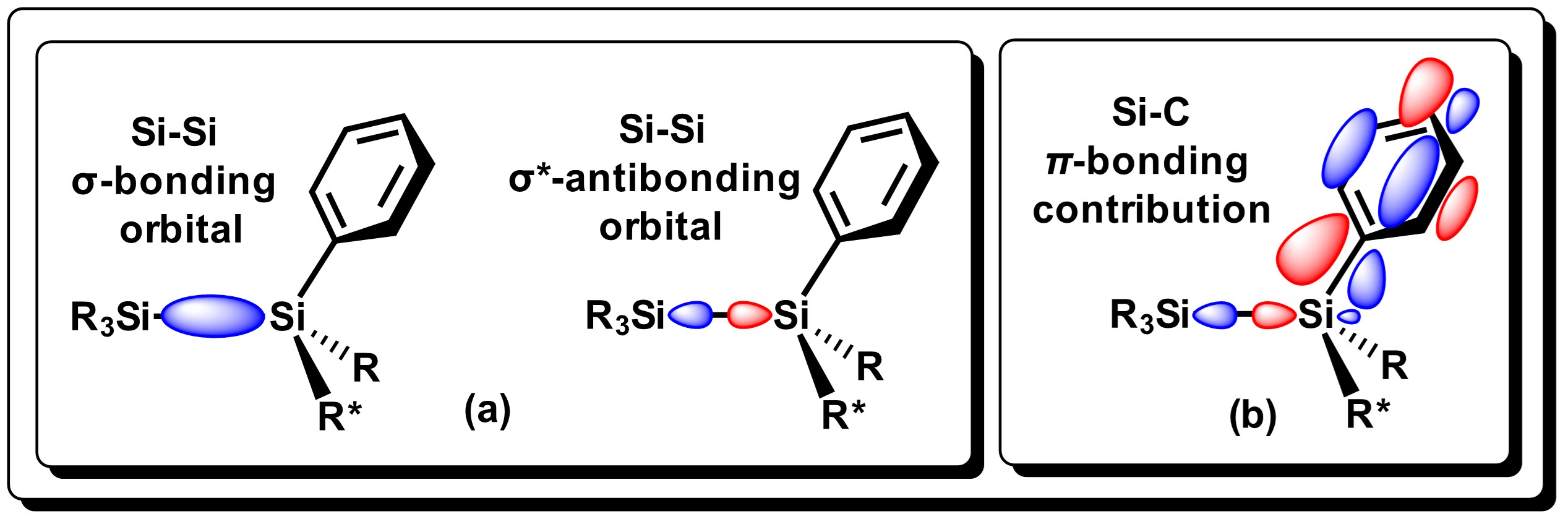
Figure 2.
Representation of the HOMO/LUMO molecular orbitals for the Si−Si bond in aryldisilanes, and the interaction of the molecular orbital. Adapted from reference
[10].
.
3. Si−H Bonds Properties
Si−H bonds properties
The Si−H bond is a polarized sigma bond which is more basic than H−H and Si−Si; hence, it behaves as a better electron-donor group. Hydrosilanes, with general formula [SiHnR4-n] are versatile starting reagents, useful for the generation of silyl-metal complexes. As with molecular hydrogen, hydrosilanes can bond directly to metals via 3c-2e non-classic interactions, with the formation of very stable compounds. These type of bonding interactions are explained via backbonding of electron density from the d-orbitals of the metal atom to the σ* molecular orbitals of the Si−H bond, which, as with the interactions of disilanes, diminishes the bond order and elongates the bond. A series of intermediates can be formed, as seen in Figure 3. Strong backdonation causes complete oxidative addition of the bond, resulting in the expected hydrido-silyl metal compounds [11]. Several complexes have been subject to theoretical studies and also determined by neutron diffraction in the solid state, where Si-H strong interactions to the metal center can be seen, like in the manganese cyclopentadienyl complex reported by McGrady et al. [12]
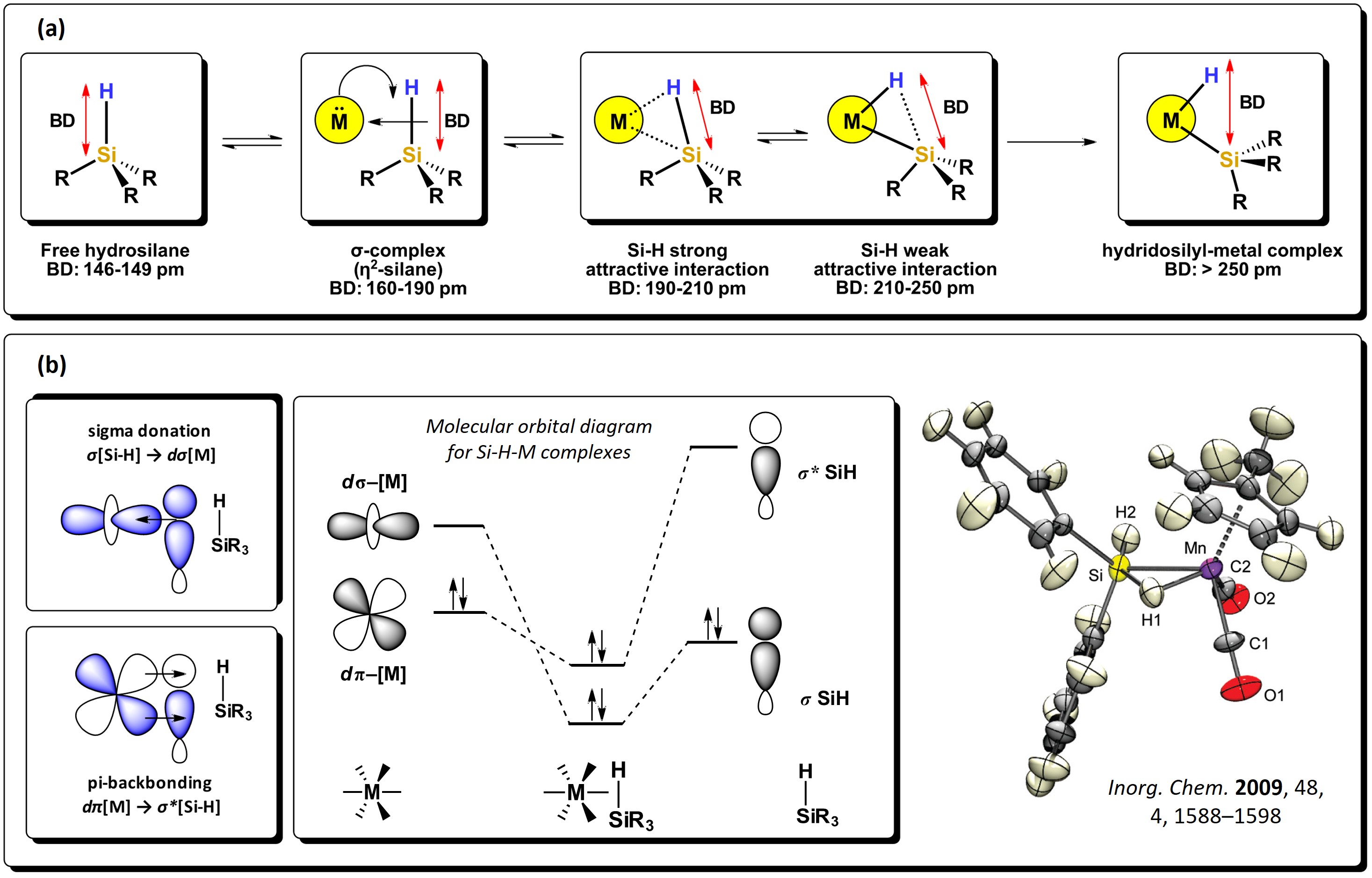
Figure 3.
Sigma donation from the Si−H bond to the d-orbital of metal atoms: (a) different stages of oxidative addition of Si-H bond to a metal and representative bond distances. (b) Schematic representation of the molecular orbitals involved in the Si−H interactions alongside an example of a sigma complex. Adapted from references
The Si–H bond distance is generally between 146 and 149 pm, about 38 pm more than for a C–H bond, also favoring the interaction between molecular orbitals of hydrosilanes with d-atomic orbitals of metal atoms. The Si−H bond, depending on the substituents attached to silicon, presents a wide range of dissociation energies. For example, for the series of hydrosilanes: F3SiH, H3SiH, Me3SiH, Et3SiH, PhH2SiH, (H3Si)H2SiH, (TMS)Me2SiH, (TMS)3SiH, a decrease in the bond dissociation energy is observed, as shown in Figure 4. This decrease is attributed to the destabilizing effect generated by the addition of silyl groups attached to the Si−H motif, which is also related to the change in electronegativity of the substituents around the Si atom. [13]
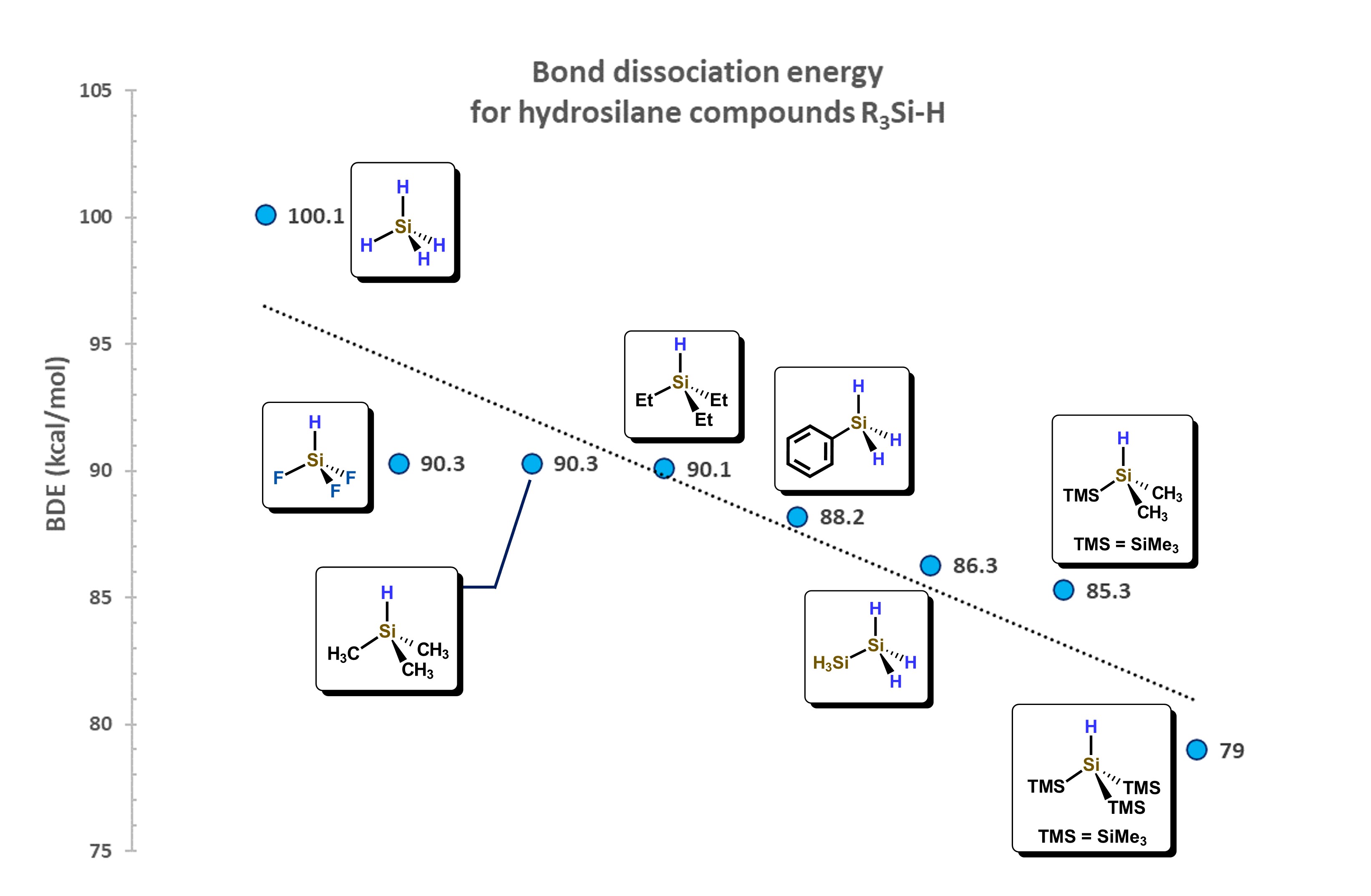
Figure 4.
Correlation of substituents bonded to Si atom and their influence in the Bond Dissociation Energy (BDE). Adapted from reference
[13]
For organosilicon compounds, important data can be obtained from coupling constants. For example, in 29Si and/or 1H NMR, couplings constants up to one bond 1JHSi are observed with values that range between 75 and 420 Hz; whereas, at two bonds, the coupling values are less than 13 Hz. In some cases, couplings at three bonds can be seen, however, these tend to be less than 8 Hz, unable to identify correctly [14]. Coupling constant is correlated to the nature of the substituents attached to the silicon atom. For the series of hydrosilanes SiH3X (X = H, F, Cl, Br and I) an increase is observed not only in the Si–H distance, but also in the H–Si–H angle caused by the effect of the substituent. Similarly, a correlation can be obtained between the coupling constants 1JHH and 1JHSi in the compounds of the type SiH3X and SiH2X2, Figure 5, and the number of X substituents on silicon. It is noteworthy that fluorosilylated compounds, as well as those with Si−N and Si−O bonds, can be analyzed in a different regression than the other compounds with Cl, Br, I, and S. On the other hand, the 1JHSi constants for fluorosilylated compounds are lower than those of the other halogenated compounds, while the 1JHH couplings are relatively higher. This dependence is related to the change in the s character of the hybrid orbital of the Si atom. In Table 2 information for simple hydrosilanes is compiled.
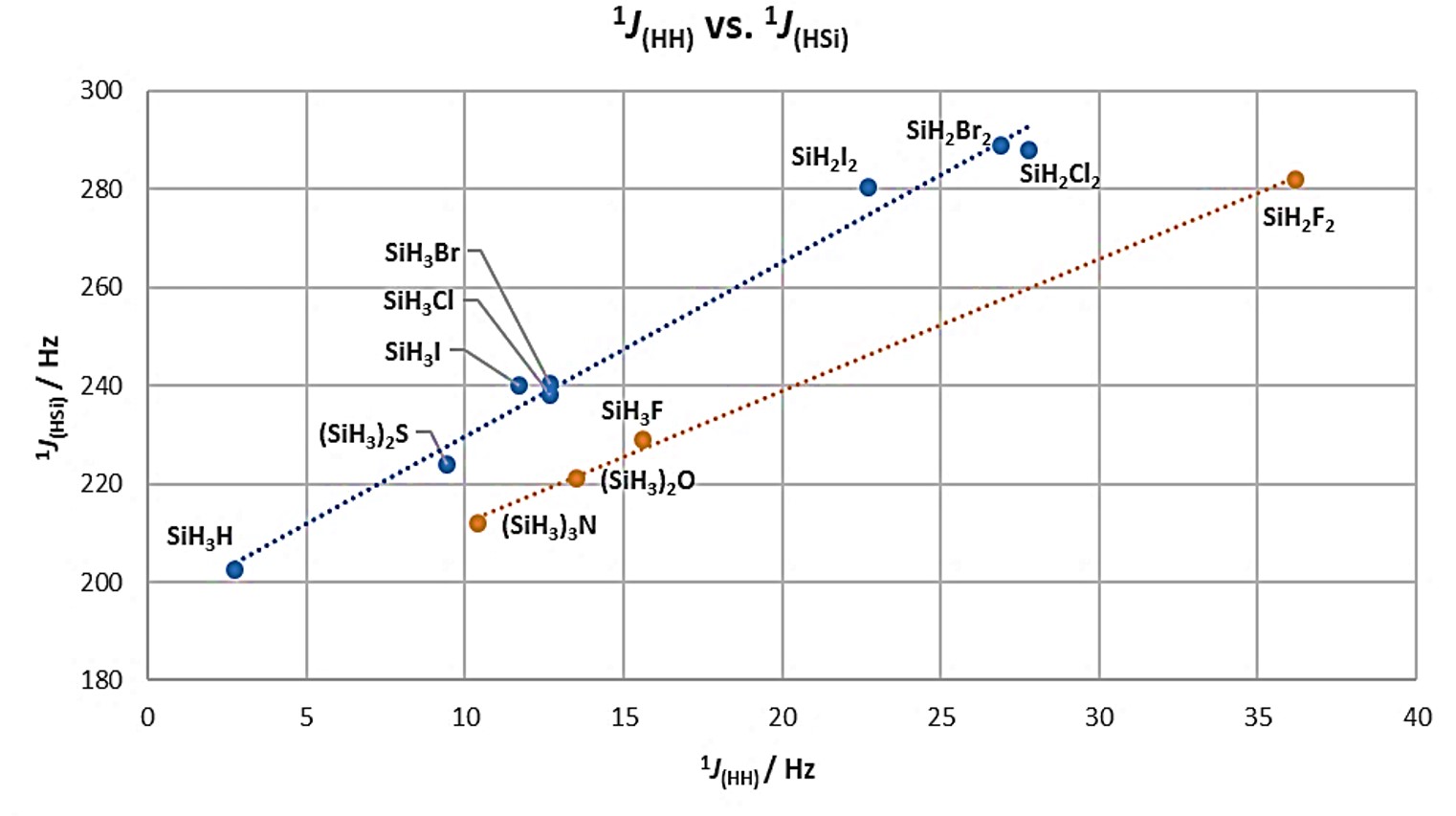
Figure 5. Scatterplot of the correlation between 1JHH and 1JHSi in a series of hydrosilanes. Adapted from references [15][16].
Table 2. Bond energies, distances and coupling constants for simple hydrosilanes. Data obtained from references [17]
| Compound | ∠H-Si-H° | r(Si-H) / pm | 1JHH (Hz) | 1JHSi (Hz) |
| SiH4 | 109.28 | 1.477 | 2.75±0.15 | 202.5±0.2 |
| SiH3F | 110.50 | 1.473 | 15.6±0.2 | 229.0±0.6 |
| SiH3Cl | 110.53 | 1.481 | 12.7±0.2 | 238.1±0.1 |
| SiH3Br | 111.7 | 1.483 | 12.7±0.5 | 240.5±0.3 |
| SiH3I | 110.54 | 1.488 | 11.7±0.3 | 240.1±0.2 |
| SiH2F2 | 114.54 | 1.467 | 36.2±0.7 | 282.0±3.0 |
| SiH2Cl2 | - | - | 27.8±0.4 | 288.0±0.4 |
| SiH2Br2 | - | - | 26.9±0.7 | 289.0±0.6 |
| SiH2I2 | - | - | 22.7±0.3 | 280.5±0.2 |
| SiHF3 | - | - | - | 381.7±1.5 |
| SiHCl3 | - | - | - | 362.9±0.2 |
| (SiH3)2O | - | - | 13.5±0.2 | 221.5±0.2 |
| (SiH3)2S | - | - | 9.43±0.13 | 224.0±0.3 |
| (SiH3)3N | - | - | 10.4±0.2 | 212.0±2.0 |
| Compound | ∠H-Si-H° | r(Si-H) / pm | 1JHH (Hz) | 1JHSi (Hz) |
| SiH4 | 109.28 | 1.477 | 2.75±0.15 | 202.5±0.2 |
| SiH3F | 110.50 | 1.473 | 15.6±0.2 | 229.0±0.6 |
| SiH3Cl | 110.53 | 1.481 | 12.7±0.2 | 238.1±0.1 |
| SiH3Br | 111.7 | 1.483 | 12.7±0.5 | 240.5±0.3 |
| SiH3I | 110.54 | 1.488 | 11.7±0.3 | 240.1±0.2 |
| SiH2F2 | 114.54 | 1.467 | 36.2±0.7 | 282.0±3.0 |
| SiH2Cl2 | - | - | 27.8±0.4 | 288.0±0.4 |
| SiH2Br2 | - | - | 26.9±0.7 | 289.0±0.6 |
| SiH2I2 | - | - | 22.7±0.3 | 280.5±0.2 |
| SiHF3 | - | - | - | 381.7±1.5 |
| SiHCl3 | - | - | - | 362.9±0.2 |
| (SiH3)2O | - | - | 13.5±0.2 | 221.5±0.2 |
| (SiH3)2S | - | - | 9.43±0.13 | 224.0±0.3 |
| (SiH3)3N | - | - | 10.4±0.2 | 212.0±2.0 |
For the series of para-substituted phenoxyhydrosilanes, SiHMe2(p-OC6H4X), with X = H, F, Cl, Br, I, NO2, there is an increase in the coupling constant to a Si-H bond as the X substituent changes, Figure 6. Notably. hydrogen substituent on the aromatic ring provides a similar result to that found for fluorine [18]. However, based on the results observed for these compounds, no specific trend can be seen for the increase in the coupling constant with the chemical shift in 29Si.
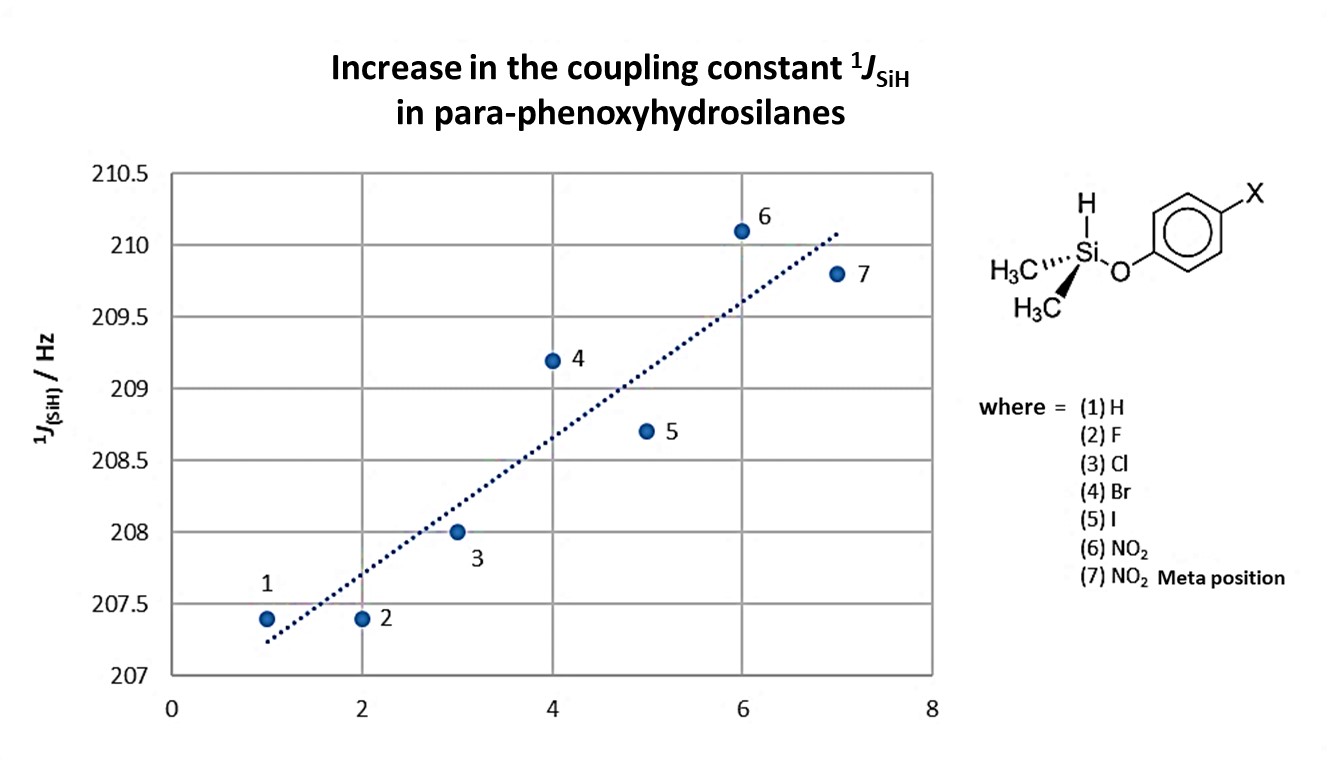
Figure 6. Increase in the 1JHSi coupling constant in a series of p-phenoxyhydrosilanes. Adapted from reference [18].
4. Reactivity of Hydrosilanes and Disilanes towards Transition Metals
Reactivity of hydrosilanes and disilanes towards transition metals
Hydrosilanes and disilanes react with a wide range of transition metal precursors. These reactions have been examined in various review articles [11]. In general, the mechanism by which silylation products are generated is through the oxidative addition of the Si−Si/Si−H bond towards a metal center. In most cases, the precursor to be used contains an electron-rich metal that, either thermally or photochemically promoted, generates vacant sites in its coordination sphere by the dissociation of neutral or anionic ligands before reaction with the respective silane. Given that in the oxidative addition process, both the oxidation number and the coordination number of the metal increase by 2 units, most precursors contain metals in low oxidation states, such as 0, +1, or +2. If there are one or more monoanionic ligands in the molecule (X = Cl-, Br-, I-, for example), a subsequent loss of HX is observed with formation of the silyl-metal product. Generally, metal precursors contain neutral ligands such as phosphines, alkenes/alkynes, H2 or CO, which provide the metal with high reactivity due to its easy dissociation in solution. The oxidative addition reactions of hydrosilanes are much more favorable than those of hydrocarbons and normally occur at room temperature. With late transition metals, such as Pt, the formation of the M-Si bond over M-C is favored as the former is a stronger bond. Although it is known that the sp3(Si−H) bond tends to be weaker than the sp3(C−H) bond, the strength of the Si−H bond depends on the substituents attached to the silicon atom causing bond strength values to be around between 75 to 100 kcal/mol [19]. Among the most commonly used hydrosilanes are those derived from PhnSiH4-n (n = 1, 2, 3), PhnMe3-nSiH (n = 1, 2), PhMeSiH2, MenCl3-nSiH (n = 1, 2), HSiR3 ( R = Et, Cl, OEt) and some simple silanes such as SiH4, H2SiCl2 and MenSiH4-n (n = 1, 2, 3), although the latter, being gases, are used imprecisely as reagents.
In general, systematic studies that show the correlation of the substituents on the silicon atom with respect of the type of product that will be generated after the reaction of a hydrosilane with a metallic precursor have not been reported. However, the diversity of products that can be obtained with small variations in the identity of the metal precursors has been reported, as illustrated in Scheme 1, which shows the reactivity of the same tertiary hydrosilane against three related Rh complexes. Products 1 to 4 vary both in geometry and in the coordination number. [20]
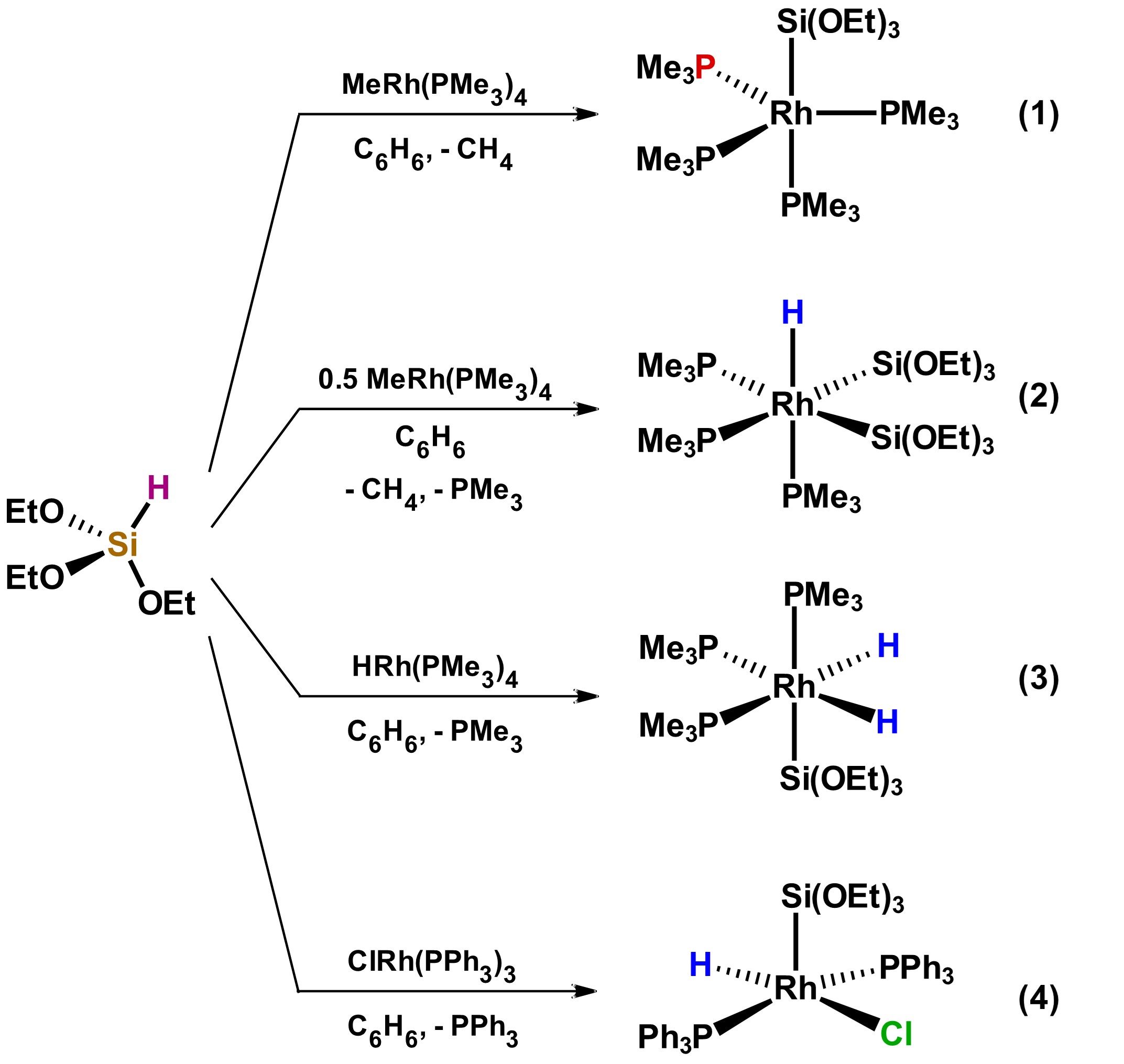
Scheme 1. Reactivity of tertiary hydrosilane HSi(OEt)3 towards different Rh precursors. Adapted from reference [20].
On the other hand, the reactivity of metal precursors that have organic fragments, M-R (generally R = Me or Ph), frequently results in their elimination in the form of RH after the oxidative addition of hydrosilane. In this sense, the reaction product replaces the organic ligand with a silyl ligand. Most of the precursors at the end of the transitional series used in this category have the methyl group as a ligand, as well as others such as phosphines. An example reaction of removal of organic fragment and its replacement by a silyl group is shown in Scheme 2. In this reaction, the addition of hydrosilane to the tungsten complex 5 forms a silyl-metal hydride species 6 that subsequently undergoes loss of methane to form the tetracoordinate compound Cp*W(CO)2(SiPh2H), 7. [21]
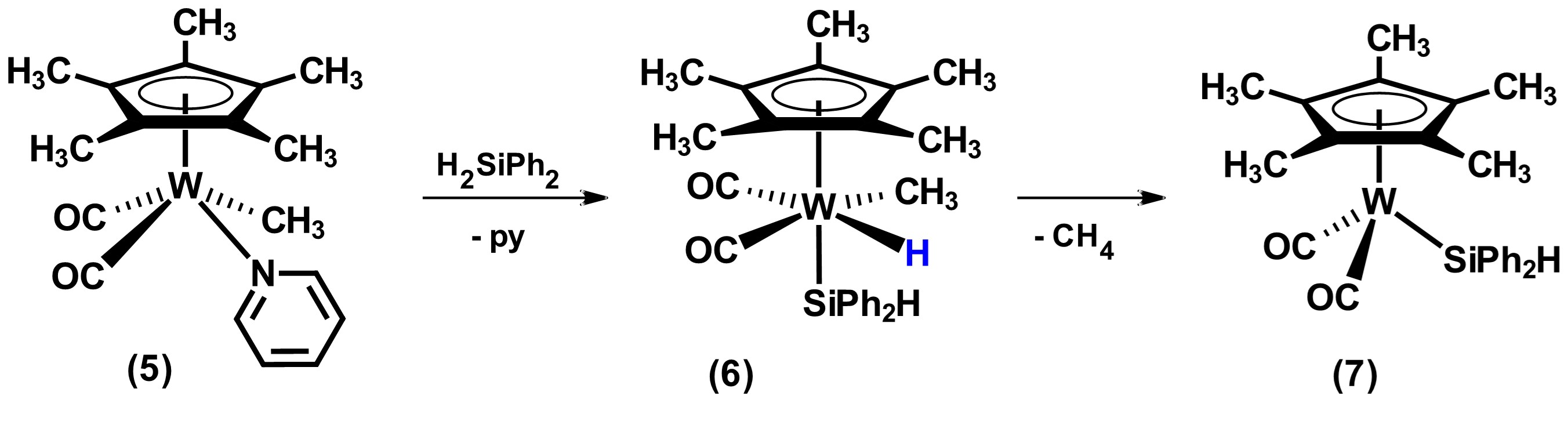
Scheme 2. Reactivity of compound [Cp*W(CO)2Me(py)] with the secondary hydrosilane H2SiPh2. Adapted from reference [21].
Another coordination mode carried out by ligands based on hydrosilanes is the one that, when incorporation of atoms such as nitrogen, oxygen and phosphorus, these are able to weakly coordinate to the metal center. Examples of hydrosilylamines and hydrosilylethers are shown in Figure 7. In these examples, chelation-assisted ligand coordination favors complex formation selectively. [22]
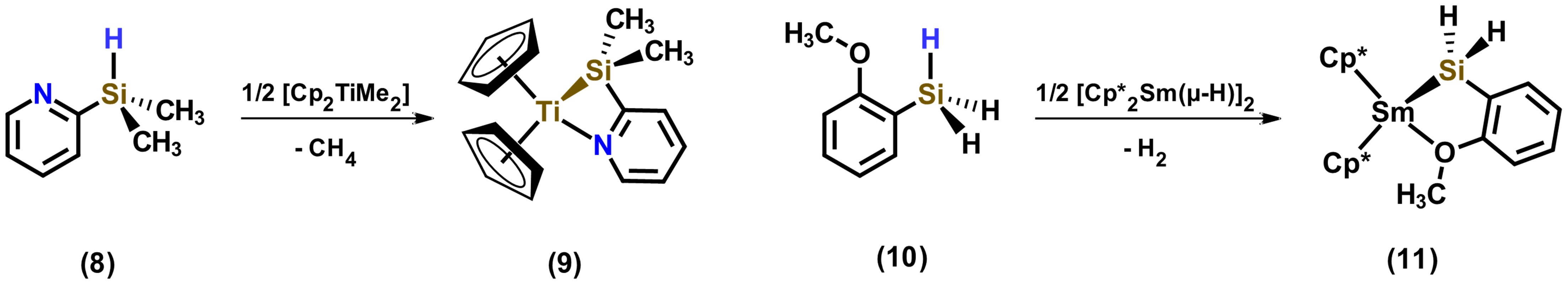
Figure 7. Hydrosilylamine and hydrosilylether as chelating ligands. Adapted from reference [22].
Reactions of hydrosilanes with metals can form species of complex nature, especially when primary and secondary hydrosilanes are used. A synthetic route that normally generates direct silylation products results from the oxidative addition of Si-Si bonds towards metals that are in low oxidation states, mainly late transition metals, such as those of the platinum group. Among the first articles on the use of disilanes towards a transition metal is the one reported by Kumada et al. in 1972, where they used the nickel(II) precursor, NiCl2(PEt3)2, in the presence of hydrodisilane, HMe2SiSiMe2H, to carry carried out the bis-silylation of di-substituted acetylene [23]. Since this first report of nickel-catalyzed bis-silylation, multiple papers on the investigation of Si-Si bond cleavage of disilanes have appeared using group 10 metals, mainly Pd and Pt. It has been generally observed that disilanes with electron-withdrawing substituents such as halogens, or in highly strained cyclic systems, possesses greater facility to cleave the bond in oxidative addition reactions towards metal precursors. Indeed, the cleavage of strained cyclic derivatives of disilanes have been proposed as ideal alternatives for the formation of metal-silyl complexes, given that the use of halogen-substituted disilanes are not always accessible. Some examples of halogen-substituted disilanes are described in Scheme 3. The compound bis(trichlorosilyl)nickel(II), 12, which has a η6 arene ligand coordinated to the metal center, is prepared by the reaction of hexachlorodisilane with nickel in the vapor phase, in the presence of toluene, which is subsequently displaced by CO generating the pentacoordinate species 13 [24]. On the other hand, it can be observed that the reaction of disilane 14 in the presence of a Pd(0) compound generates product 16 where both silyl groups remain in a trans fashion, highly unusual due to the strong trans influence exerted by the silicon atom. [25]
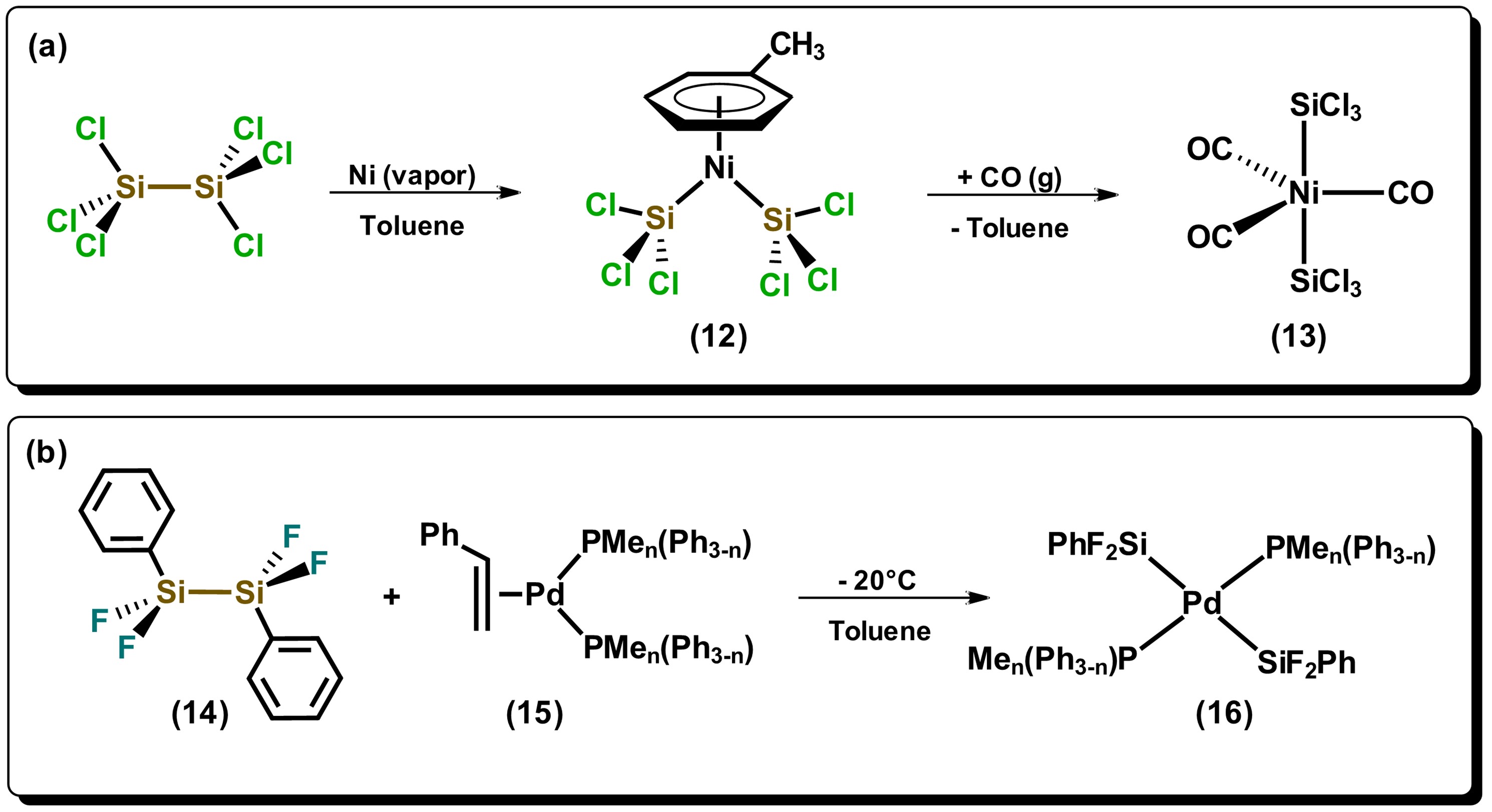
Scheme 3. Reactivity of disilanes towards electron-rich metals. Adapted from references [24][25].
5. Conclusions
Conclusions
As stated above, the chemistry of hydrosilanes and disilanes is vast. However, an understanding of the properties of these compounds and their trends associated with the change of substituents around the silicon atom remains poorly explored. The efforts published in the literature highlight the importance of the research of hydrosilanes and disilanes as important raw materials in the elaboration of complex molecules with high value for industry, academia, among others. The Si-H bond in hydrosilanes continues to generate countless reports in the literature, while the information on Si-Si bond cleavage remains less investigated. Because of the high reactivity of the Si-H bond, several issues in the elaboration of specific products are encountered, while the directionality of the Si-Si bond cleavage offers advantages as disilanes are more stable under ambient conditions.
References
- Chatt, J.; Eaborn, C.; Ibekwe S. Silicon, a ligand atom of exceptionally high trans-effect. Chem. Commun. (London) 1966, 19, 700-701, 10.1039/C19660000700.
- Armitage, D.A.. Organosilanes; Pergamon: Oxford, 1982; pp. 1-203.
- Anderson, R.; Goff, J.; Imamura, A.; Kimble, E.; Lockwood, G.; Matisons, J.; Pan, Y.; Reinert, M.. Silicon Compounds: Silanes and Silicones. A Survey of Properties and Chemistry.; 3rd Edition: Morrisville, 2013; pp. 1-608.
- Sakurai, H.; Kamiyama, Y.; Nakadaira, Y. Chemistry of organosilicon compounds. 79. Novel [.sigma. + .pi.]reactions of hexaorganodisilanes with acetylenes catalyzed by palladium complexes. J. Am. Chem. Soc. 1975, 97, 931-932, 10.1021/ja00837a061.
- Kaftory, M.; Kapon, M.; Botoshansky, M. . The Structural Chemistry of Organosilicon Compounds, from The Chemistry of Organic Silicon Compounds; Rappoport, Zvi; Apeloig, Yitzhak , Eds.; John Wiley & Sons, Ltd: USA, 1998; pp. 181-265.
- Bock, H.; Meuret, J.; Ruppert, K. Sterically overcrowded or charge perturbed molecules: XXIII. Hexakis(trimethylsilyl)disilane: Structure and photoelectron spectrum of a sterically overcrowded molecule. J. Organomet. Chem. 1993, 445, 19-28, 10.1016/0022-328X(93)80181-A.
- Gilman, H; Lichtenwalter, G.D. Cleavage of Symmetrically Substituted Disilanes by Lithium in Tetrahydrofuran. J. Am. Chem. Soc. 1958, 80, 608-611, 10.1021/ja01536a025.
- Gilman, H.; Lichtenwalter, G.D.; Wittenberg, D. Cleavage Studies of Disilanes by Silyllithium Compounds. J. Am. Chem. Soc. 1959, 81, 5320-5322, 10.1021/ja01529a019.
- Tamao, K.; Kawachi, A.; Nakagawa, Y.; Ito, Y. Electronic spectra of (amino)(phenyl)disilanes. J. Organomet. Chem. 1994, 473, 29-34, 10.1016/0022-328X(94)80102-9.
- Däschlein, C.; Strohmann, C. The competition between Si–Si and Si–C cleavage in functionalised oligosilanes: their reactivity with elemental lithium. Dalton Trans. 2010, 39, 2062-2069, 10.1039/B920846A.
- Corey, J.Y. Reactions of Hydrosilanes with Transition Metal Complexes and Characterization of the Products. Chem. Rev. 2011, 111, 863-1071, 10.1021/cr900359c.
- McGrady, G.S.; Sirsch, P.; Chatterton, N.P.; Ostermann, A.; Gatti, C.; Altmannshofer, S.; Herz, V.; Eickerling, G.; Scherer, W. Nature of the Bonding in Metal-Silane σ-Complexes. Inorg. Chem. 2009, 48, 1588–1598, 10.1021/ic8019777.
- Kanabus-Kaminska, J. M.; Hawari, J. A.; Griller, D.; Chatgilialoglu, C. Reduction of silicon-hydrogen bond strengths. J. Am. Chem. Soc. 1987, 109, 5267–5268, 10.1021/ja00251a035.
- Gupta, R.; Lechner, M.. Chemical shifts and coupling constants for Silicon-29; Springer: Switzerland , 2008; pp. 1-476.
- A. Rastelli, S.A. Pozzoli Hybridization, structure and 29Si-H coupling constants in methyl and phenyl derivatives of silane. J. Molec. Struct. 1973, 18, 463-469, 10.1016/0022-2860(73)85097-5.
- Ebsworth, E. A. V.; Frankiss, S. G. Nuclear magnetic resonance studies of silicon hydrides and derivatives. Part 3.—Chemical shifts and spin-spin coupling constants in monomethylsilyl compounds. Trans. Faraday Soc. 1963, 59, 1518-1524, 10.1039/TF9635901518.
- M.A. Jensen The prediction of 29Si-H and 13C-H coupling constants in substituted silanes and methanes. J. Organomet. Chem. 1968, 11, 423-427, 10.1016/0022-328X(68)80066-X.
- Larin, M. F.; Dubinskaya, É. I.; Voronkov, M. G.; Pestunovich, V. A. 1H and1H-29Si NMR spectra of dimethylaroxysilanes. Russ. Chem. Bull. 1981, 30, 1552–1554, 10.1007/BF00952214.
- Ding, L; Marshall, P. Does alkyl substitution affect the silicon-hydrogen bond strength in silane? Kinetic studies of the reactions of atomic chlorine and bromine with trimethylsilane and an ab initio investigation. J. Am. Chem. Soc. 1992, 114, 5754–5758, 10.1021/ja00040a041.
- Aizenberg, M.; , Ott, J; , Elsevier, C.J.; Milstein, D. Rh(I) and Rh(III) silyl PMe3 complexes. Syntheses, reactions and 103Rh NMR spectroscopy. J. Organomet. Chem. 1998, 551, 81-92, 10.1016/S0022-328X(97)00433-6.
- Sakaba, H.; Tsukamoto, M.; Hirata, T.; Kabuto, C.; Horino, H. Synthesis, Structure, and Silylene Exchange Reaction of Base-Stabilized Hydrido(silylene)tungsten Complexes and Rearrangement of Hydrosilyl(pyridine)tungsten Complexes to the Base-Stabilized Hydrido(silylene) Complexes via 1,2-Hydrogen Migration. J. Am. Chem. Soc 2000, 122, 11511–11512, 10.1021/ja993279w.
- Castillo, I.; Tilley, T.D. Mechanistic Aspects of Samarium-Mediated σ-Bond Activations of Arene C−H and Arylsilane Si−C Bonds. J. Am. Chem. Soc. 2001, 123, 10526–10534, 10.1021/ja011472w.
- Okinoshima, H.; Yamamoto, K.; Kumada, M. Dichlorobis(triethylphosphine)nickel(II) as a catalyst for reactions of sym-tetramethyldisilane with unsaturated hydrocarbons. Novel synthetic routes to 1-silacyclopentadienes and 1,4-bis(dimethylsilyl)2-butenes. J. Am. Chem. Soc. 1972, 94, 9263–9264, 10.1021/ja00781a066.
- Groshens, T. J.; Klabunde, K.J. Carbonyl compounds of cobalt(II) and nickel(II). Reversible arene-carbon monoxide exchange and similarities to analogous η6-arene complexes. J. Organomet. Chem. 1983, 259, 337-343, 0.1016/0022-328X(83)87184-8.
- Ozawa, F.; Sugawara, M.; Hayashi, T. A New Reactive System for Catalytic Bis-Silylation of Acetylenes and Olefins. Organometallics 1994, 13, 3237–3243, 10.1021/om00020a042.