Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Yi-Han Chiu.
The mechanisms by which immune systems identify and destroy tumors, known as immunosurveillance, have been discussed for decades. However, several factors that lead to tumor persistence and escape from the attack of immune cells in a normal immune system have been found. In the process known as immunoediting, tumors decrease their immunogenicity and evade immunosurveillance. Furthermore, tumors exploit factors such as regulatory T cells, myeloid-derived suppressive cells, and inhibitory cytokines that avoid cytotoxic T cell (CTL) recognition.
- immunogenicity
- tumor antigens
- immunoediting
1. The Sentry of the Immune System: Immunosurveillance
1.1. Tumor Recognition and Rejection by the Immune System
T lymphocytes are essential in antitumor immunity, such as in the surveillance, detection, and destruction of neoplastic cells [1]. CTLs are the most potent effectors and are considered significant drivers in antitumor effects [4][2], and they are activated by dendritic cells (DCs). DCs initiate and maintain the antitumor T cell immunity [5][3]. During tumor initiation, the dying tumor cells release danger signals, such as molecules called damage-associated molecular patterns (DAMPs). Upon sensing these signals and capturing tumor cells, DCs undergo maturation, migrate to the draining lymph nodes (dLNs), and present the tumor antigens onto major histocompatibility complex I (MHC I) for presentation to CD8+ T cells [20][4]. These activate the antigen-specific CTLs, which naïve CD8+ T cells differentiate into CTLs and memory CD8+ T cells. The educated CTLs can recognize the antigenic targets expressed on the tumors and secrete cytokines, IFN-γ, perforin, and granzyme, thus performing tumor lytic functions [6,7][5][6]. In addition to priming naïve CD8+ T cells, DCs interact with memory T cells and induce their differentiation into the tumor sites [21][7]. Furthermore, DCs produce IL-12, which triggers IFN-γ release from the CTLs, which in turn enhances CTL activation and functions [22][8]. The close interaction between DCs and CTLs suggests tight T cell–DC cross-talk in the tumor microenvironment (TME).
CTLs exerting antitumor effects rely on the help of other immune cells, such as DCs and helper CD4+ T cells. Helper CD4+ T cells play a prominent role in maintaining CD8+ cytolytic responses and preventing CTL exhaustion [4,23][2][9]. Interestingly, the help signals originating from the CD40 ligand (CD40L) on the helper CD4+ T cells stimulate CD40 on the DCs, which deliver CD4+ T cell-derived help signals to CD8+ T cells and elicit CTL responses [24][10]. During the first antigen priming step, naïve CD4+ T cells and CD8+ T cells are separately activated by different populations of DCs in the peripheral lymph nodes [25[11][12][13],26,27], where MHC I-expressing cDC1 usually interacts with CD8+ T cells. In contrast, MHC II-expressing cDC2 interacts with CD4+ T cells. Next, in the second priming step, CD4+ T cells and CD8+ T cells interact with the same cDC1, and the help signal occurs [28,29][14][15]. cDC1 engages in cognate interaction with pre-activated CD4+ T cells, which optimizes the cDC1 to relay signals for the differentiation of effector T cells and memory CTLs to pre-activated CD8+ T cells [30][16]. In summary, antigen-specific (pre-activated) CD4+ T cells assist the pre-activated CD8+ T cells in the differentiation into effector T or memory T cells. Helper CD4+ T cells dictate the quality of the CTL differentiation and promote the expansion of antigen-specific CTLs by cytokine signals. CD4+ T cells promote the development of antigen-specific CTLs through the amplification of IL-12 and IL-15 production induced by IFN-γ in DCs [31,32][17][18]. Furthermore, IL-2 signaling promotes CD8 proliferation [33,34][19][20], and the expression of IL-2 receptor α-chain (IL-2Rα) by recently primed CD8+ T cells depends on CD4+ T cells [35][21]. In addition to the assistant roles of CD4+ T cells, the cytolytic activities in antitumor effects of CD4+ T cells have been proposed. CD4+ T cells produce IFN-γ and express granzymes and perforin to kill tumors [36,37][22][23]. Recent studies using RNA sequencing in human cancers have reported cytotoxic characteristics of CD4+ T cells, which express cytolytic molecules such as granzymes, perforin, and granulysin in the tumors and circulation of cancer patients [38,39,40,41][24][25][26][27].
Natural killer (NK) cells are also essential effectors in tumor immunosurveillance. For example, NK cells can be activated by MHC class I polypeptide-related sequence A (MICA) and MICB, which are expressed on tumors [2][28]. NK cells directly kill tumor cells by releasing perforin and granzymes [3][29] or trigger cell apoptosis through the ligation of cell death receptor-mediated pathways (FasL/Fas) [42][30]. Healthy cells express high levels of MHC I, which ligates to the killer immunoglobulin-like family inhibitory receptors (KIRs) on NK cells. However, tumors will downregulate MHC I expression to evade CTL-mediated cytotoxicity and simultaneously activate NK cells due to the decreased inhibitory signaling [42,43][30][31] (Figure 1).
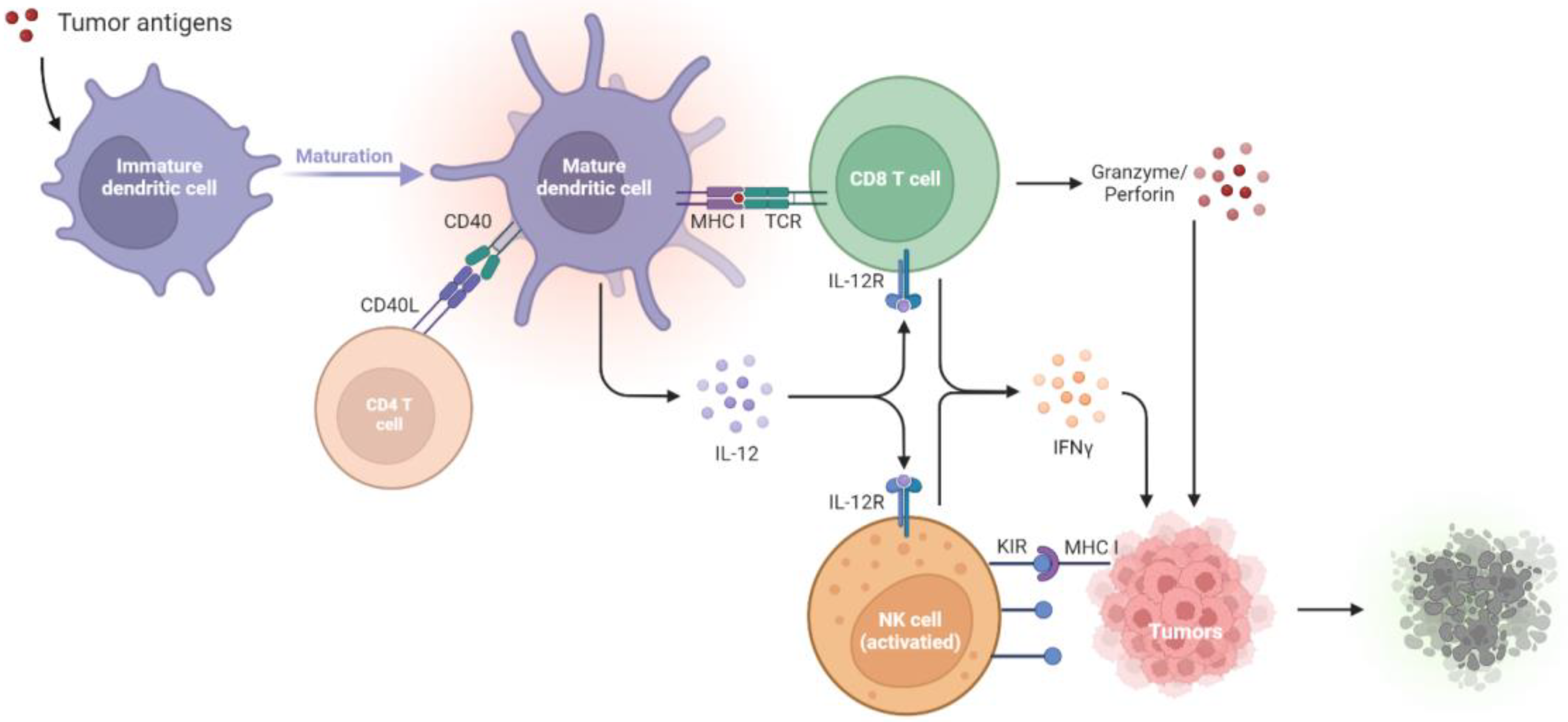
Figure 1. Tumor suppression by the immune systems during immunosurveillance. Once tumor antigens are captured, immature DCs undergo maturation. The mature DCs present the antigens onto MHC I molecules for presentation to CD8+ T cells. The CD40L on the CD4+ T cell stimulates CD40 on the DCs, which delivers help signals to CD8+ T cells. With the CD40–CD40L interaction, the amplification of IL-12 in DCs promotes the development of antigen-specific CD8+ T cells and thus increases the IFN-γ, granzyme, and perforin production to kill tumors. NK cells will be activated by the diminished expression of MHC I on tumors, which relieves the KIR inhibitory signaling and activates the NK cell toxicity towards tumors. CD40L, CD40 ligand; DCs, dendritic cells; IL-12R, IL-12 receptor; IFN-γ, interferon-γ; KIR, killer immunoglobulin-like receptor; MHC I, major histocompatibility complex class I; NK cells, natural killer cells; TCR, T cell receptor.
1.2. Failure of Immunosurveillance Enables Cancer Progression
Accumulating evidence indicates the increased risk of tumor development in immunosuppressed patients, suggesting the crucial antitumor role of intact immunity. Mice with a deficiency in adaptive immunity provide a practical animal model for testing cancer immunosurveillance. In one study, mice with severe combined immunodeficiency (SCID) exhibited impaired differentiation of both T and B lymphocytes, and thus 15% of these mice developed T cell lymphomas [44][32]. In another study, RAG2−/− mice, another kind of mice without B and T cells, developed spontaneous intestinal adenomas (50%), intestinal adenocarcinomas (35%), and lung cancers (15%) at the age of 15–16 months [45][33]. In addition to adaptive immunity, NK cells also manifested cancer immunoediting by producing IFN-γ and induced M1 macrophages.
Mice with a deficiency in effector cells, such as T cells and NK cells, usually develop spontaneous cancers with aging. In STAT1-deficient mice, the JAK/STAT1 signaling pathway is down-regulated and thus inhibits type I and II IFN production. This leads to the increased formation of mammary carcinomas [46][34]. Mice lacking death-inducing molecule tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) [47][35], or with the inactivation of the Fas-mediated cell death pathway by Fas or FasL mutation, have accelerated hematological malignancies [48][36]. Decreased cytokine production and/or antigen presentation in gene-deficient mice, including Perforin−/− (lack perforin) [49][37], Ifng−/− (lack IFN-γ) [50][38], Perforin−/−Ifng−/− (lack perforin and IFN-γ) [50][38], Perforin−/−B2m−/− (lack perforin and MHC class I expression) [51][39], and Lmp2−/− (defective MHC class I antigen presentation) [52][40], facilitates tumor growth. Due to the decreased IFN-γ secretion, IL-12 and IL-18, two IFN-γ-inducing cytokines, are down-regulated. A previous study found that, compared with wild-type mice, neither IL-12 nor IL-18 deficient mice exhibited increased incidence of tumor development [50][38]. These results suggest that immune effector cells, cytokines, and their related pathways are essential for the regression of tumor development in immunosurveillance. The following findings support the same conclusion. Due to the presence of the constitutively expressing oncogene, Kras, and inhibiting tumor suppressor, p53, tumors growing in immunocompetent mice were retarded compared to those in immunocompromised mice.
2. Immunoediting: How Tumors Hijack Host Immunity and Establish a Favorable Microenvironment
Tumors escape from the immune system via several mechanisms, including the formation of regulatory cells, reduced immune recognition, and production of suppressive cytokines. The immune system fails to restrict tumor development; as a result, tumors evade immune recognition (due to diminished tumor antigens and immunogenicity) and release suppressive cytokines. The regulatory immune cells, Tregs, MDSCs, dysfunctional DCs, and M2 macrophages also express immune regulatory factors, such as IDO and arginase, to construct a tumor microenvironment (TME) that enhances tumor progression and dampens T cell functions. In this escape phase, the balance is toward tumor progression, with the infiltrations of inhibitory cells, cytokines, and factors. The results are that the immune system is incapable of inhibiting tumor progression. By generating the appropriate TME via the mechanisms listed below, tumors dampen the immune responses, supporting tumor growth and even metastasis [8][41].
2.1. Recruitment of Regulatory Cells
2.1.1. Regulatory T Cells
Immune suppression mediated by the regulatory cells and other suppressive factors in the TME is a major mechanism by which tumors avoid attacks from the immune system. CD4+CD25+FoxP3+ tumor-derived regulatory T cells (Tregs) play central roles in immune suppression. Studies also show that different cytokines, such as IL-10 and TGF-β produced by the tumors, trigger the conversion of CD4+ T cells into suppressive Tregs [53][42]. Tregs then secret IL-10, TGF-β, and IL-35 to downregulate antitumor immunity, suppress the antigen presentation by DCs, and decrease the tumor-specific CTLs [54][43]. Meanwhile, since IL-2 is essential to T cell activation and maintenance, these regulatory T cells compete with effector T cells by largely consuming IL-2 [33,34][19][20]. Tregs can also kill the CTLs by the secretion of perforin and granzyme, leading to osmotic lysis and apoptosis, just as CTLs and NK cells do to tumor cells. Furthermore, Tregs interfere with the memory T cells by repressing their effector and proliferation activities through the upregulation of CTLA-4 ligands [55][44]. The aforementioned evidence supports the mediation of the comprehensive suppression of antitumor immunity by Tregs.
2.1.2. Myeloid-Derived Suppressive Cells
Both Tregs and myeloid-derived suppressive cells (MDSCs) facilitate their own expansion by the overexpression of CD73 [56[45][46],57], TGF-β [58][47], and indoleamine 2, 3-dioxygenase (IDO) [59,60,61][48][49][50]. In addition, MDSCs may promote the recruitment of Tregs by producing CCR5 ligands, CCL3, CCL4, and CCL5 [62][51]. MDSCs express a high level of inducible nitric oxide (iNO), which produces nitric oxide (NO) [63,64][52][53]. NO inhibits the JAK/STAT5 pathway and/or suppresses the antigen presentation from DCs, leading to the suppressive proliferation of effector T cells [64,65][53][54]. Furthermore, MDSC-derived NO also triggers the effector T cell apoptosis [66][55] and reduces E-selectin expression on endothelial cells, which hampers the migration of T cells to the tumor sites [67,68][56][57]. MDSCs also secrete a high level of arginase 1 (ARG1), which promotes Treg expansion [69][58] and depletes the L-arginine, a conditionally essential amino acid in effector T cell functions. These actions lead to the dysregulation of effector T cells and the propagation of Tregs [70][59].
2.1.3. Tumor-Associated Macrophages
Among the regulatory cell types, tumor-associated macrophages (TAMs) are the most abundant population in TME [71][60]. There are two polarization states of TAMs: classically activated M1 and alternatively activated M2 subtypes [72][61]. M2 macrophages produce several anti-inflammatory cytokines, such as IL-4, IL-10, IL-13, vascular endothelial growth factor (VEGF), and TGF-β, to inhibit immune systems and promote tumor progression [73,74,75][62][63][64]. Therefore, CD11b+F4/80+ macrophages having an M2 phenotype are considered regulatory (or “bad”) macrophages [76][65], which are essential for tumor growth and metastasis. [77][66]. Furthermore, M2 macrophages can support tumor-related vasculature by accumulating vascular endothelial cells [78][67]. M2 polarization is induced by several factors, one of which is a colony-stimulating factor 1 (CSF1). CSF1 plays a fundamental role in pro-angiogenesis and tumor burden increase [79][68], revealing M2 macrophages as the efficient cancer development enabler within the TME [77][66]. In fact, M2 participates in the angiogenesis cascade, which consists of a series of pro-tumor functions, including degradation of extracellular matrix (ECM), endothelial cell proliferation, and migration [80][69]. M2-related enzymes promote ECM deposition and the proteolysis of collagens. The degraded collagen fragments may stimulate M2, which could further re-arrange stroma and enhance the angiogenesis activities [81][70]. Therefore, M2 macrophages are immune cells that promote tumors by releasing suppressive cytokines, triggering tumor angiogenesis, and reorganizing the ECM in the TME.
2.2. Defective Antigen Presentation
2.2.1. Manipulation of the DC Lineage
Defective antigen presentation is another fundamental mechanism by which tumors evade immune surveillance. Immunogenic tumors will be recognized and destroyed by immunosurveillance. However, tumors evolve multiple skills for escaping from immune recognition, including the suppression of DC functions and downregulation of MHC-I expression [82][71]. DCs play essential roles in the initiation of antitumor T cell immunity [5][3]. Among the DC subsets, cDC1s (c: conventional) are the most important [83][72], for the abundance of cDC1s in the TME is associated with T cell infiltration and overall survival in cancer patients [20,84][4][73]. cDC1s are recruited to tumor regions by chemokines released by the tumors, such as CCL4 and CCL5 [85,86][74][75]. After taking up tumor cells, cDC1s will mature in the tumor sites and migrate to the dLNs to process tumor antigens onto MHC-I for presentation to CD8+ T cells [20][4]. These processes result in the activation of antigen-specific CTLs. Thus, cDC1s participate in antitumor activities and serve as targets for tumors to escape from the immune system. Tumors prevent the accumulation of cDC1s by activating their β-catenin signaling pathways, thereby decreasing the infiltration of cDC1s and T cells [85,86][74][75]. Furthermore, tumors release high levels of prostaglandin E2 (PGE2) [87][76], VEGF [88[77][78],89], IL-6 [90][79], IL-10 [91][80], and TGF-β [92][81] to suppress DC maturation and differentiation. Thus, tumors can decrease their antigenicity by manipulating DC functions, which become tolerogenic and immunosuppressive phenotypes.
2.2.2. Sabotage of the Machinery of Antigen Presentation
The downregulation of tumor antigenicity, such as MHC class I loss, is a well-studied characteristic of immune evasion from tumor-specific cytolytic effects. Tumor antigenicity is sculpted during immunoediting in an immunocompetent host. Some of these changes include antigen depletion [93][82], gene alterations in MHC-I and B2M [94][83], and modulation of other antigen processing and presentation machinery (APM). Several TAAs are the by-products during tumorigenesis, which indicates that these TAAs are not necessarily functional for tumor growth. The loss of such antigens in tumors can prevent immune predation [82][71]. MHC-I and B2M mutations are usually found in tumors [94][83], and they lead to reduced surface expression of MHC-I [95][84]. Diminished B2M expression is correlated to a cold immune environment, with low T cell infiltration [93][82]. Tumors evading immune surveillance by downregulation of APM components are also widely described. Deficiency in proteasome subunits [96][85], transporters associated with antigen processing (TAP) proteins [97][86], and Tapasin [98][87] result in inadequate antigen presentation, thereby escaping CTL recognition. In conclusion, the downregulation of tumor antigens and antigen-presenting processes leads to enhanced tumor growth and metastasis as the CTLs fail to identify the targets on the tumor cells.
2.3. Immune Suppressive Mediators
Tumors can also inhibit host immunity by releasing regulatory cytokines. Within the TME, tumor-induced cytokines and inflammation facilitate cancer development [99,100][88][89]. Several tumor-promoting cytokines are regulated by nuclear factor-κB (NF-κB), a central orchestrator of inflammation. Previous research has revealed that the inactivation of NF-κB in immune cells decreases the expression of proinflammatory cytokines and thus relieves the tumor burden [101][90]. These results indicate that NF-κB-mediated cytokines, including IL-1 [102][91], IL-6 [103][92], IL-8 [104][93], and TNF-α [102][91], are highly correlated to tumorigenesis. For example, IL-6 interacts with the receptor JAK and induces STAT-3 activation [103][92], which triggers oncogenes such as myeloid cell leukemia-1 (MCL-1) and upregulates proliferative genes, Cyclin-D1, in tumor cells [105][94]. In addition, TGF-β is one of the most important contributors to tumor growth. TGF-β is released by the cancer cells, Tregs, fibroblasts, and other types of cells within the TME. The elevated TGF-β levels inhibit effector T cell differentiation, promote Treg proliferation, and dampen DC functions [106][95]. Furthermore, TGF-β enhances the epithelial-to-mesenchymal transition (EMT), synergistically triggering tumor metastasis with IL-6 via the overactivation of JAK/STAT signaling pathways [107][96]. Therefore, TGF-β is usually regarded as a chief mediator within the immunosuppressive factors [108][97].
The overproduction of PGE2 [87][76], VEGF [88[77][78],89], IL-6 [90][79], IL-10 [91][80], and TGF-β [92][81] from tumors will inhibit DC differentiation. The undifferentiated DCs fail to appropriately present antigens and are unable to educate T cells. Immunosuppressive enzymes, such as IDO and arginase, also cause tumor progression through the induction of T-cell tolerance and tumor cell proliferation. IDO impairs CTL functions through the downregulation of the T cell receptor ζ chain and enhances Treg generation [109,110][98][99]. Elevated intertumoral IDO expression is correlated to T cell dysfunction, such as decreased levels of granzyme B in tumor-infiltrating CD8+ T cells [111][100], impaired degranulation of γδ T cells [112][101], and upregulation of PD-1 and PD-L1 ligands [113,114][102][103]. Tumor-infiltrating myeloid cells produce high levels of arginase, which inhibit T cell receptor expression, dampen antigen-specific T cell antitumor immunity, and induce Treg proliferation [115,116][104][105].
2.4. Deletion of Tumor-Specific CTLs
Tumors and several immunosuppressive cells trigger T cell apoptosis by Fas and Fas ligand (FasL) signaling pathways. FasL is expressed by tumors and can induce apoptosis of Fas-expressing antitumor CTLs [117][106]. FasL is reported to be expressed on the tumoral endothelium but not normal vasculature [118][107], suggesting that FasL-expressing endothelial cells in tumors induce Fas-mediated apoptosis in CTLs. Furthermore, these phenomena have only been observed in CTLs, not Tregs [119,120][108][109]. FasL-expressing MDSCs also result in the deletion of CTLs [121,122][110][111].
The high expression of CD70 and PD-L1 on the surfaces of tumors also mediates T cell death. The TNF receptor family member CD27 is usually expressed on T cells [123][112], but its ligand, CD70, is overexpressed in some tumors [124][113]. The dysregulation of the CD70–CD27 axis within the TME is associated with immunosuppression [125][114]. One explanation is that the over-activation of CD27 can lead to T-cell deletion, for Siva, a pro-apoptotic protein, can interact with CD27 through a caspase-dependent pathway [126][115]. Therefore, tumors can induce immunosuppression by promoting T-cell apoptosis via the expression of CD70.
PD-L1 and PD-1 induce immune suppression and facilitate tumor growth by induction of T cell apoptosis. PD-L1 is highly expressed in numerous types of cancers, including numerous solid tumors and hematological cancers [127][116]. When tumor-expressing PD-L1 combines with PD-1 on the T cells, SHP-1/2 are recruited to the C-terminal PD-1. This causes the de-phosphorylation of several vital signal transducers, including ZAP70, CD3δ, and PI3K pathways, and thus inhibits T cell proliferation, reduces cytokine production, and triggers T cell apoptosis [127,128][116][117].
References
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416.
- van der Leun, A.M.; Thommen, D.S.; Schumacher, T.N. CD8(+) T cell states in human cancer: Insights from single-cell analysis. Nat. Rev. Cancer 2020, 20, 218–232.
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24.
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336.
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761.
- Nolz, J.C. Molecular mechanisms of CD8(+) T cell trafficking and localization. Cell Mol. Life Sci. 2015, 72, 2461–2473.
- Enamorado, M.; Iborra, S.; Priego, E.; Cueto, F.J.; Quintana, J.A.; Martínez-Cano, S.; Mejías-Pérez, E.; Esteban, M.; Melero, I.; Hidalgo, A.; et al. Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat. Commun. 2017, 8, 16073.
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199.
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367.
- Ridge, J.P.; Di Rosa, F.; Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 1998, 393, 474–478.
- Bedoui, S.; Heath, W.R.; Mueller, S.N. CD4(+) T-cell help amplifies innate signals for primary CD8(+) T-cell immunity. Immunol. Rev. 2016, 272, 52–64.
- Calabro, S.; Liu, D.; Gallman, A.; Nascimento, M.S.; Yu, Z.; Zhang, T.T.; Chen, P.; Zhang, B.; Xu, L.; Gowthaman, U.; et al. Differential Intrasplenic Migration of Dendritic Cell Subsets Tailors Adaptive Immunity. Cell Rep. 2016, 16, 2472–2485.
- Gerner, M.Y.; Casey, K.A.; Kastenmuller, W.; Germain, R.N. Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J. Exp. Med. 2017, 214, 3105–3122.
- Eickhoff, S.; Brewitz, A.; Gerner, M.Y.; Klauschen, F.; Komander, K.; Hemmi, H.; Garbi, N.; Kaisho, T.; Germain, R.N.; Kastenmüller, W. Robust Anti-viral Immunity Requires Multiple Distinct T Cell-Dendritic Cell Interactions. Cell 2015, 162, 1322–1337.
- Hor, J.L.; Whitney, P.G.; Zaid, A.; Brooks, A.G.; Heath, W.R.; Mueller, S.N. Spatiotemporally Distinct Interactions with Dendritic Cell Subsets Facilitates CD4+ and CD8+ T Cell Activation to Localized Viral Infection. Immunity 2015, 43, 554–565.
- Groom, J.R.; Richmond, J.; Murooka, T.T.; Sorensen, E.W.; Sung, J.H.; Bankert, K.; von Andrian, U.H.; Moon, J.J.; Mempel, T.R.; Luster, A.D. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 2012, 37, 1091–1103.
- Wang, J.C.; Livingstone, A.M. Cutting edge: CD4+ T cell help can be essential for primary CD8+ T cell responses in vivo. J. Immunol. 2003, 171, 6339–6343.
- Greyer, M.; Whitney, P.G.; Stock, A.T.; Davey, G.M.; Tebartz, C.; Bachem, A.; Mintern, J.D.; Strugnell, R.A.; Turner, S.J.; Gebhardt, T.; et al. T Cell Help Amplifies Innate Signals in CD8(+) DCs for Optimal CD8(+) T Cell Priming. Cell. Rep. 2016, 14, 586–597.
- Spolski, R.; Li, P.; Leonard, W.J. Biology and regulation of IL-2: From molecular mechanisms to human therapy. Nat. Rev. Immunol. 2018, 18, 648–659.
- Carmenate, T.; Ortíz, Y.; Enamorado, M.; García-Martínez, K.; Avellanet, J.; Moreno, E.; Graça, L.; León, K. Blocking IL-2 Signal In Vivo with an IL-2 Antagonist Reduces Tumor Growth through the Control of Regulatory T Cells. J. Immunol. 2018, 200, 3475.
- Obar, J.J.; Molloy, M.J.; Jellison, E.R.; Stoklasek, T.A.; Zhang, W.; Usherwood, E.J.; Lefrançois, L. CD4+T cell regulation of CD25 expression controls development of short-lived effector CD8+ T cells in primary and secondary responses. Proc. Natl. Acad. Sci. USA 2010, 107, 193–198.
- Xie, Y.; Akpinarli, A.; Maris, C.; Hipkiss, E.L.; Lane, M.; Kwon, E.K.; Muranski, P.; Restifo, N.P.; Antony, P.A. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 2010, 207, 651–667.
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650.
- Zhang, L.; Yu, X.; Zheng, L.; Zhang, Y.; Li, Y.; Fang, Q.; Gao, R.; Kang, B.; Zhang, Q.; Huang, J.Y.; et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018, 564, 268–272.
- Zhang, Q.; He, Y.; Luo, N.; Patel, S.J.; Han, Y.; Gao, R.; Modak, M.; Carotta, S.; Haslinger, C.; Kind, D.; et al. Landscape and Dynamics of Single Immune Cells in Hepatocellular Carcinoma. Cell 2019, 179, 829–845.e20.
- Oh, D.Y.; Kwek, S.S.; Raju, S.S.; Li, T.; McCarthy, E.; Chow, E.; Aran, D.; Ilano, A.; Pai, C.S.; Rancan, C.; et al. Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020, 181, 1612–1625.e13.
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20.
- Amin, P.J.; Shankar, B.S. Sulforaphane induces ROS mediated induction of NKG2D ligands in human cancer cell lines and enhances susceptibility to NK cell mediated lysis. Life Sci. 2015, 126, 19–27.
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7.
- Smyth, M.J.; Cretney, E.; Kelly, J.M.; Westwood, J.A.; Street, S.E.; Yagita, H.; Takeda, K.; van Dommelen, S.L.; Degli-Esposti, M.A.; Hayakawa, Y. Activation of NK cell cytotoxicity. Mol. Immunol. 2005, 42, 501–510.
- Handgretinger, R.; Lang, P.; André, M.C. Exploitation of natural killer cells for the treatment of acute leukemia. Blood 2016, 127, 3341–3349.
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530.
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111.
- Chan, S.R.; Vermi, W.; Luo, J.; Lucini, L.; Rickert, C.; Fowler, A.M.; Lonardi, S.; Arthur, C.; Young, L.J.T.; Levy, D.E.; et al. STAT1-deficient mice spontaneously develop estrogen receptor α-positive luminal mammary carcinomas. Breast Cancer Res. 2012, 14, R16.
- Zerafa, N.; Westwood, J.A.; Cretney, E.; Mitchell, S.; Waring, P.; Iezzi, M.; Smyth, M.J. Cutting Edge: TRAIL Deficiency Accelerates Hematological Malignancies. J. Immunol. 2005, 175, 5586–5590.
- Davidson, W.F.; Giese, T.; Fredrickson, T.N. Spontaneous development of plasmacytoid tumors in mice with defective Fas-Fas ligand interactions. J. Exp. Med. 1998, 187, 1825–1838.
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760.
- Street, S.E.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134.
- Street, S.E.; Hayakawa, Y.; Zhan, Y.; Lew, A.M.; MacGregor, D.; Jamieson, A.M.; Diefenbach, A.; Yagita, H.; Godfrey, D.I.; Smyth, M.J. Innate immune surveillance of spontaneous B cell lymphomas by natural killer cells and gammadelta T cells. J. Exp. Med. 2004, 199, 879–884.
- Hayashi, T.; Faustman, D.L. Development of spontaneous uterine tumors in low molecular mass polypeptide-2 knockout mice. Cancer. Res. 2002, 62, 24–27.
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146.
- Paluskievicz, C.M.; Cao, X.; Abdi, R.; Zheng, P.; Liu, Y.; Bromberg, J.S. T Regulatory Cells and Priming the Suppressive Tumor Microenvironment. Front. Immunol. 2019, 10, 2453.
- Sullivan, J.A.; Tomita, Y.; Jankowska-Gan, E.; Lema, D.A.; Arvedson, M.P.; Nair, A.; Bracamonte-Baran, W.; Zhou, Y.; Meyer, K.K.; Zhong, W.; et al. Treg-Cell-Derived IL-35-Coated Extracellular Vesicles Promote Infectious Tolerance. Cell Rep. 2020, 30, 1039–1051.e35.
- Li, C.; Jiang, P.; Wei, S.; Xu, X.; Wang, J. Regulatory T cells in tumor microenvironment: New mechanisms, potential therapeutic strategies and future prospects. Mol. Cancer 2020, 19, 116.
- Morello, S.; Pinto, A.; Blandizzi, C.; Antonioli, L. Myeloid cells in the tumor microenvironment: Role of adenosine. Oncoimmunology 2016, 5, e1108515.
- Ohta, A.; Kini, R.; Ohta, A.; Subramanian, M.; Madasu, M.; Sitkovsky, M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front. Immunol. 2012, 3, 190.
- Ghiringhelli, F.; Puig, P.E.; Roux, S.; Parcellier, A.; Schmitt, E.; Solary, E.; Kroemer, G.; Martin, F.; Chauffert, B.; Zitvogel, L. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J. Exp. Med. 2005, 202, 919–929.
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401.
- Moon, Y.W.; Hajjar, J.; Hwu, P.; Naing, A. Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J. Immunother. Cancer 2015, 3, 51.
- Jitschin, R.; Braun, M.; Büttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760.
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J. Immunol. 2012, 189, 5602–5611.
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.C.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int. J. Cancer. 2014, 134, 2853–2864.
- Markowitz, J.; Wang, J.; Vangundy, Z.; You, J.; Yildiz, V.; Yu, L.; Foote, I.P.; Branson, O.E.; Stiff, A.R.; Brooks, T.R.; et al. Nitric oxide mediated inhibition of antigen presentation from DCs to CD4+ T cells in cancer and measurement of STAT1 nitration. Sci. Rep. 2017, 7, 15424.
- Bingisser, R.M.; Tilbrook, P.A.; Holt, P.G.; Kees, U.R. Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J. Immunol. 1998, 160, 5729–5734.
- Wang, Z.; Jiang, J.; Li, Z.; Zhang, J.; Wang, H.; Qin, Z. A myeloid cell population induced by Freund adjuvant suppresses T-cell-mediated antitumor immunity. J. Immunother. 2010, 33, 167–177.
- Gehad, A.E.; Lichtman, M.K.; Schmults, C.D.; Teague, J.E.; Calarese, A.W.; Jiang, Y.; Watanabe, R.; Clark, R.A. Nitric oxide-producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J. Investig. Dermatol. 2012, 132, 2642–2651.
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962.
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer. Res. 2008, 68, 5439–5449.
- Rodríguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191.
- Dehne, N.; Mora, J.; Namgaladze, D.; Weigert, A.; Brüne, B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr. Opin. Pharmacol. 2017, 35, 12–19.
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends. Immunol. 2015, 36, 229–239.
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237.
- van Kempen, L.C.; Ruiter, D.J.; van Muijen, G.N.; Coussens, L.M. The tumor microenvironment: A critical determinant of neoplastic evolution. Eur. J. Cell Biol. 2003, 82, 539–548.
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355.
- Jeannin, P.; Paolini, L.; Adam, C.; Delneste, Y. The roles of CSFs on the functional polarization of tumor-associated macrophages. FEBS J. 2018, 285, 680–699.
- Spring, H.; Schüler, T.; Arnold, B.; Hämmerling, G.J.; Ganss, R. Chemokines direct endothelial progenitors into tumor neovessels. Proc. Natl. Acad. Sci. USA 2005, 102, 18111–18116.
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.W.; Vunjak-Novakovic, G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014, 35, 4477–4488.
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology 2013, 2, e26968.
- Butoi, E.; Gan, A.M.; Tucureanu, M.M.; Stan, D.; Macarie, R.D.; Constantinescu, C.; Calin, M.; Simionescu, M.; Manduteanu, I. Cross-talk between macrophages and smooth muscle cells impairs collagen and metalloprotease synthesis and promotes angiogenesis. Biochim. Biophys. Acta 2016, 1863, 1568–1578.
- Hu, W.Q.; Fang, M.; Zhao, H.L.; Yan, S.G.; Yuan, J.P.; Peng, C.W.; Yang, G.F.; Li, Y.; Li, J.D. Tumor invasion unit in gastric cancer revealed by QDs-based in situ molecular imaging and multispectral analysis. Biomaterials 2014, 35, 4125–4132.
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312.
- Böttcher, J.P.; Reis e Sousa, C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends. Cancer 2018, 4, 784–792.
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652.
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235.
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141.
- Hangai, S.; Ao, T.; Kimura, Y.; Matsuki, K.; Kawamura, T.; Negishi, H.; Nishio, J.; Kodama, T.; Taniguchi, T.; Yanai, H. PGE2 induced in and released by dying cells functions as an inhibitory DAMP. Proc. Natl. Acad. Sci. USA 2016, 113, 3844–3849.
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103.
- Ohm, J.E.; Carbone, D.P. VEGF as a mediator of tumor-associated immunodeficiency. Immunol. Res. 2001, 23, 263–272.
- Park, S.J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K.; et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 173, 3844–3854.
- Yang, A.S.; Lattime, E.C. Tumor-induced interleukin 10 suppresses the ability of splenic dendritic cells to stimulate CD4 and CD8 T-cell responses. Cancer Res. 2003, 63, 2150–2157.
- Papaspyridonos, M.; Matei, I.; Huang, Y.; do Rosario Andre, M.; Brazier-Mitouart, H.; Waite, J.C.; Chan, A.S.; Kalter, J.; Ramos, I.; Wu, Q.; et al. Id1 suppresses anti-tumour immune responses and promotes tumour progression by impairing myeloid cell maturation. Nat. Commun. 2015, 6, 6840.
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485.
- Castro, A.; Ozturk, K.; Pyke, R.M.; Xian, S.; Zanetti, M.; Carter, H. Elevated neoantigen levels in tumors with somatic mutations in the HLA-A, HLA-B, HLA-C and B2M genes. BMC. Med. Genom. 2019, 12, 107.
- Shukla, S.A.; Rooney, M.S.; Rajasagi, M.; Tiao, G.; Dixon, P.M.; Lawrence, M.S.; Stevens, J.; Lane, W.J.; Dellagatta, J.L.; Steelman, S.; et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat. Biotechnol. 2015, 33, 1152–1158.
- Boulpicante, M.; Darrigrand, R.; Pierson, A.; Salgues, V.; Rouillon, M.; Gaudineau, B.; Khaled, M.; Cattaneo, A.; Bachi, A.; Cascio, P.; et al. Tumors escape immunosurveillance by overexpressing the proteasome activator PSME3. Oncoimmunology 2020, 9, 1761205.
- Tabassum, A.; Samdani, M.N.; Dhali, T.C.; Alam, R.; Ahammad, F.; Samad, A.; Karpiński, T.M. Transporter associated with antigen processing 1 (TAP1) expression and prognostic analysis in breast, lung, liver, and ovarian cancer. J. Mol. Med. 2021, 99, 1293–1309.
- Shionoya, Y.; Kanaseki, T.; Miyamoto, S.; Tokita, S.; Hongo, A.; Kikuchi, Y.; Kochin, V.; Watanabe, K.; Horibe, R.; Saijo, H.; et al. Loss of tapasin in human lung and colon cancer cells and escape from tumor-associated antigen-specific CTL recognition. Oncoimmunology 2017, 6, e1274476.
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41.
- Quinn, K.M.; Kartikasari, A.E.R.; Cooke, R.E.; Koldej, R.M.; Ritchie, D.S.; Plebanski, M. Impact of age-, cancer-, and treatment-driven inflammation on T cell function and immunotherapy. J. Leukoc. Biol. 2020, 108, 953–965.
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296.
- Zinatizadeh, M.R.; Schock, B.; Chalbatani, G.M.; Zarandi, P.K.; Jalali, S.A.; Miri, S.R. The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 2021, 8, 287–297.
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis. Rev. 2010, 29, 273–283.
- Manna, S.K.; Ramesh, G.T. Interleukin-8 induces nuclear transcription factor-kappaB through a TRAF6-dependent pathway. J. Biol. Chem. 2005, 280, 7010–7021.
- Leslie, K.; Lang, C.; Devgan, G.; Azare, J.; Berishaj, M.; Gerald, W.; Kim, Y.B.; Paz, K.; Darnell, J.E.; Albanese, C.; et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006, 66, 2544–2552.
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940.
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 2011, 9, 1658–1667.
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin. Cancer Biol. 2015, 35, S185–S198.
- Chen, W.; Liang, X.; Peterson, A.J.; Munn, D.H.; Blazar, B.R. The indoleamine 2,3-dioxygenase pathway is essential for human plasmacytoid dendritic cell-induced adaptive T regulatory cell generation. J. Immunol. 2008, 181, 5396–5404.
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol. 2020, 11, 531491.
- Li, R.; Zhang, H.; Cao, Y.; Liu, X.; Chen, Y.; Qi, Y.; Wang, J.; Yu, K.; Lin, C.; Liu, H.; et al. Lauren classification identifies distinct prognostic value and functional status of intratumoral CD8(+) T cells in gastric cancer. Cancer Immunol. Immunother. 2020, 69, 1327–1336.
- Jonescheit, H.; Oberg, H.H.; Gonnermann, D.; Hermes, M.; Sulaj, V.; Peters, C.; Kabelitz, D.; Wesch, D. Influence of Indoleamine-2,3-Dioxygenase and Its Metabolite Kynurenine on γδ T Cell Cytotoxicity against Ductal Pancreatic Adenocarcinoma Cells. Cells 2020, 9, 1140.
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116.
- Gide, T.N.; Allanson, B.M.; Menzies, A.M.; Ferguson, P.M.; Madore, J.; Saw, R.P.M.; Thompson, J.F.; Long, G.V.; Wilmott, J.S.; Scolyer, R.A. Inter- and intrapatient heterogeneity of indoleamine 2,3-dioxygenase expression in primary and metastatic melanoma cells and the tumour microenvironment. Histopathology 2019, 74, 817–828.
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I Production in the Tumor Microenvironment by Mature Myeloid Cells Inhibits T-Cell Receptor Expression and Antigen-Specific T-Cell Responses. Cancer Res. 2004, 64, 5839–5849.
- Wakkach, A.; Fournier, N.; Brun, V.; Breittmayer, J.P.; Cottrez, F.; Groux, H. Characterization of dendritic cells that induce tolerance and T regulatory 1 cell differentiation in vivo. Immunity 2003, 18, 605–617.
- Barnhart, B.C.; Legembre, P.; Pietras, E.; Bubici, C.; Franzoso, G.; Peter, M.E. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004, 23, 3175–3185.
- Yu, J.S.; Lee, P.K.; Ehtesham, M.; Samoto, K.; Black, K.L.; Wheeler, C.J. Intratumoral T cell subset ratios and Fas ligand expression on brain tumor endothelium. J. Neurooncol. 2003, 64, 55–61.
- Bajou, K.; Peng, H.; Laug, W.E.; Maillard, C.; Noel, A.; Foidart, J.M.; Martial, J.A.; DeClerck, Y.A. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell 2008, 14, 324–334.
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615.
- Horton, B.L.; Williams, J.B.; Cabanov, A.; Spranger, S.; Gajewski, T.F. Intratumoral CD8(+) T-cell Apoptosis Is a Major Component of T-cell Dysfunction and Impedes Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 14–24.
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.-F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent antigen at vaccination sites induces tumor-specific CD8⁺ T cell sequestration, dysfunction and deletion. Nat. Med. 2013, 19, 465–472.
- Wasiuk, A.; Testa, J.; Weidlick, J.; Sisson, C.; Vitale, L.; Widger, J.; Crocker, A.; Thomas, L.J.; Goldstein, J.; Marsh, H.C.; et al. CD27-Mediated Regulatory T Cell Depletion and Effector T Cell Costimulation Both Contribute to Antitumor Efficacy. J. Immunol. 2017, 199, 4110–4123.
- Jacobs, J.; Deschoolmeester, V.; Zwaenepoel, K.; Rolfo, C.; Silence, K.; Rottey, S.; Lardon, F.; Smits, E.; Pauwels, P. CD70: An emerging target in cancer immunotherapy. Pharmacol. Ther. 2015, 155, 1–10.
- Flieswasser, T.; Van den Eynde, A.; Van Audenaerde, J.; De Waele, J.; Lardon, F.; Riether, C.; de Haard, H.; Smits, E.; Pauwels, P.; Jacobs, J. The CD70-CD27 axis in oncology: The new kids on the block. J. Exp. Clin. Cancer Res. 2022, 41, 12.
- Py, B.; Slomianny, C.; Auberger, P.; Petit, P.X.; Benichou, S. Siva-1 and an alternative splice form lacking the death domain, Siva-2, similarly induce apoptosis in T lymphocytes via a caspase-dependent mitochondrial pathway. J. Immunol. 2004, 172, 4008–4017.
- Ostrand-Rosenberg, S.; Horn, L.A.; Haile, S.T. The programmed death-1 immune-suppressive pathway: Barrier to antitumor immunity. J. Immunol. 2014, 193, 3835–3841.
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10.
More