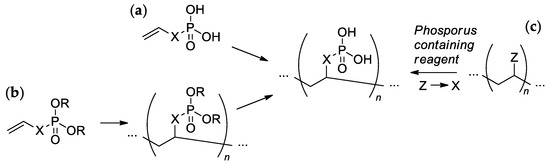
2. Design and Synthesis of Sidechain PCPAs
2.1. Synthetic Approaches to Sidechain PCPAs: An Overview
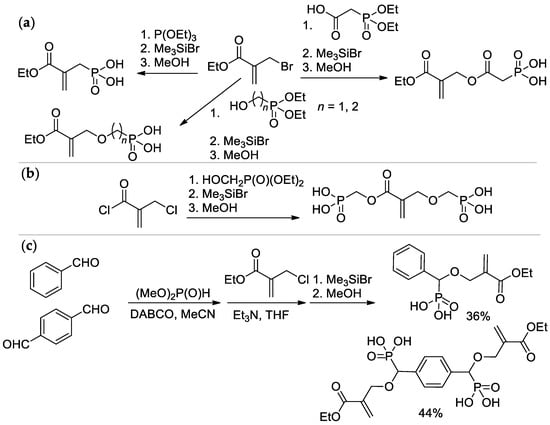
Most of the synthetic approaches to sidechain PCPAs use (co)polymerization of the phosphorus-containing vinyl monomers (
Scheme 1a,b). The alternative approach is based on the post-modification of the (co)polymers (
SScheme 1c
heme 1c).
Scheme 1. The common synthetic approaches to sidechain PCPAs: (a) polymerization of unsaturated phosphoric or phosphinic acids; (b) polymerization of unsaturated phosphoesters followed by hydrolysis; (c) phosphorylation of polymers.
2.2. Phosphorus Containing Vinyl Monomers
Vinyl monomers, containing phosphonate or phosphate fragments, are definitely starting compounds suitable for the synthesis of sidechain PCPAs, and that was the reason why
rwe
searchers chose to devote a separate section to these compounds. The synthesis and chemistry of the phosphorus-containing vinyl monomers were discussed previously in the review of Macarie and Ilia
[4][7], outlining poly(vinylphosphonic acid) and its derivatives, and in the review of David and Coll.
[5][10], devoted to phosphonate vinyl monomers and polymers. Monomers and PCPAs containing fluoro substituents in the main polymer chain were mentioned in the review of Améduri and coll.
[6][9]. A representative list of vinyl monomers suitable for the preparation of adhesive (co)polymers for dentistry was presented in the work of Moszner, Salz, and Zimmermann
[7][5].
2.2.1. Vinylphosphonic Acid and Related Compounds
Vinylphosphonic acid (VPA) CH
2=CHP(O)(OH)
2 represents the simplest monomer for the synthesis of sidechain PCPAs
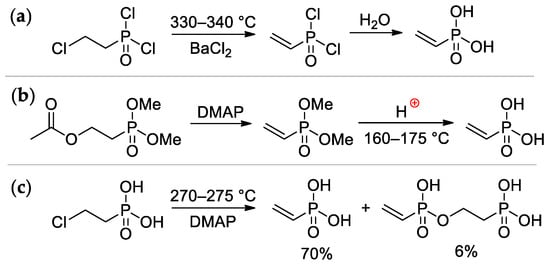
[4][7]. It was obtained for the first time by Kabachnik and Medved in 1959 via dehydrochlorination of ClCH
2CH
2P(O)Cl
2 (330–340 °C, BaCl
2) followed by hydrolysis of CH
2=CHP(O)Cl
2 intermediate
[8][15] (
Scheme 2a); in turn, ClCH
2CH
2P(O)Cl
2 was prepared by the reaction of PCl
3 with oxirane, rearrangement of the resulting P(OCH
2CH
2Cl)
3 to ClCH
2CH
2P(O)(OCH
2CH
2Cl)
2, followed by its reaction with thionyl chloride
[9][16]. Another method was based on the reaction of AcOCH
2CH
2P(O)(OMe)
2 with DMAP, followed by acidic hydrolysis of the CH
2=CHP(O)(OMe)
2 intermediate
[10][17]. (
Scheme 2b). The cost-effective synthetic approach was based on a readily available starting compound ClCH
2CH
2P(O)(OH)
2 (used as a growth regulator for plants), its pyrolysis was accompanied by the formation of the bis(phosphonate) side product
[11][18] (
SScheme 2c
heme 2c). The use of microwave irradiation in the pyrolysis of ClCH
2CH
2P(O)(OH)
2 proved ineffective
[12][19]. Vinylphosphonic acid is a colorless, low-melting solid (M.p. 36 °C)
[13][20].
Scheme 2. The synthesis of vinylphosphonic acid: (
a) from (2-chloroethyl)dichlorophopshonate
[8][15]; (
b) from AcOCH
2CH
2P(O)(OMe)
2 [10][17]; (
c) based on commercially available(2-chloroethyl) phopshonic acid
[11][18].
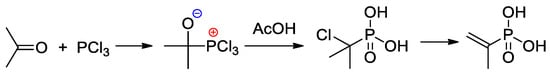
An efficient method of the synthesis of 2-propenylphosphonic acid involves the nucleophilic addition of PCl
3 to acetone followed by the reaction with AcOH and subsequent dehydrochlorination
[14][21] (
Scheme 3).
Scheme 3. The synthesis of 2-propenylphosphonic acid
[14][21].
2.2.2. Phosphorylated Acrylates and Related Compounds
The high reactivity of the esters of acrylic and methacrylic acids in polymerization and their greater synthetic availability in comparison with VPA and VPA esters are the reasons for researchers’ continued interest in vinyl monomers of this type. Among others, derivatives of (2-hydroxyethyl)methacrylate (HEMA) deserve special mention due to their synthetic availability.
Different synthetic approaches to phosphate- and phosphonate-functionalized acrylates are presented and discussed below. Evidently, phosphate or phosphonate groups can be introduced as a part of the alkoxy fragment of (meth)acrylate (the easiest and most affordable way,
Scheme 4), but functionalization of the methyl group of methacrylate also leads to prospective monomers (
Scheme 5).
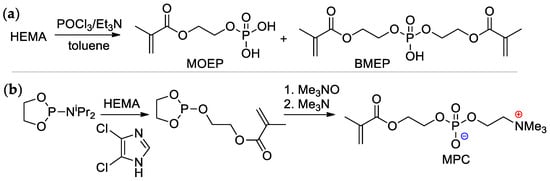
Scheme 4. Synthesis of (meth)acrylates containing –P(O)(OH)
2 group in alkoxy fragment: (
a) phosphoryl chloride based approach; (
b) 1,3,2-dioxaphospholan-2-amine based approach
[15][16][17][22,23,24].
Scheme 5. Synthesis of phosphonated methacrylates via functionalization of the methyl group: (a) based on ethyl 2-(bromomethyl)acrylate; (b) from 2-(chloromethyl)acryloyl chloride; (c) via condensation of aldehydes with dimethyl phosphonate.
2-(Methacryloyloxy)ethyl phosphate (MOEP) and bis[2-(methacryloyloxy)ethyl]phosphate (BMEP) were synthesized by the reaction of HEMA with POCl
3, followed by hydrolysis, the yields of the products depended on the HEMA/POCl
3 ratio
[15][16][22,23] (
Scheme 4a). Direct condensation of HEMA with phosphorylcholine is accompanied by polymerization, and the synthesis of methacryloyl phosphorylcholine (MPC) was carried out using the ‘phosphoramidite’ route with an overall yield of 37%
[17][24] (
Scheme 4b).
The Michaelis–Arbuzov reaction of α-bromo ethyl methacrylate with P(OEt)
3 with subsequent hydrolysis resulted in the formation of (2-(ethoxycarbonyl)allyl)phosphonic acid
[15][18][22,25], while its acylation by 2-(diethoxyphosphoryl)acetic acid and hydrolysis allowed to obtain (2-((2-(ethoxycarbonyl)allyl)oxy)-2-oxoethyl)phosphonic acid
[19][26]; alkylation and hydrolysis lead to ether-linked acrylic phosphonates
[20][27] (
Scheme 5a). Interaction of 2-(chloromethyl)acryloyl chloride with hydroxymethyl diethyl phosphonate afforded bis(phosphinic acid)
[18][25] (
Scheme 5b). Synthesis of aromatic acrylic phosphonates was carried out by the Pudovik reaction, starting from benzaldehyde or terephthaldialdehyde
[15][20][22,27] (
Scheme 5c).
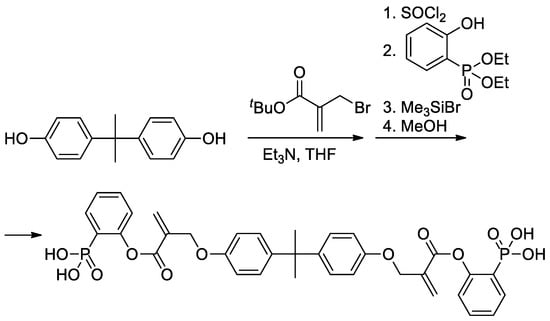
As can be seen in
Scheme 5b, both alkoxy and methyl groups of methacrylate can be affected by modification. The applicability of this approach was also demonstrated by Avci and coll et al., who used the example of the synthesis of Bisphenol A-based bis(phosphonic acid) (
Scheme 6)
[21][28].
Scheme 6. Synthesis of bis(phosphonic acid) from Bisphenol A
[21][28].
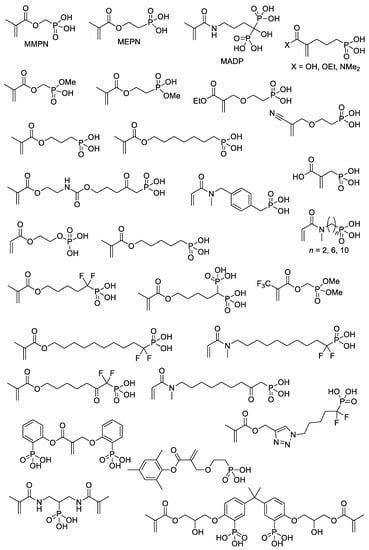
With the use of common synthetic approaches (
Scheme 4 and
Scheme 5) and other equally simple reactions, dozens of –P(O)(OH)
2-functionalized acrylates were synthesized
[15][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][22,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43] (
Scheme 7). An important note is that an incomplete list of acrylates, presented in
Scheme 7, can be further expanded by introducing dialkyl phosphonate since deprotection with the formation of PCPAs can be easily carried out after polymerization
[37][38][44,45]. However, a number of acrylate monomers containing –P(O)(OR)
2 groups and polymers obtained on their basis were not studied in the synthesis of PCPAs
[39][40][46,47].
Scheme 7. The structures of the (meth)acrylates containing –P(O)(OH)
2 fragments
[15][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][22,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43].
2.2.3. Other Phosphorus Containing Monomers
Fluorinated polymers represent a separate group of materials due to specific surface characteristics and high thermal and chemical stability. In a recent review by Améduri et al.
[6][9], different synthetic strategies for fluorinated (co)polymers containing phosphorus groups are discussed, with the consideration of –P(O)(OH)
2-functionalized monomers and polymers. However, the synthesis of such type compounds was described in a single article by Yamabe et al. who developed a complex and time-consuming method for the synthesis of perfuorovinyloxy-substituted perfuoroalkylphosphonic acid derivatives CF
2=CFO(CF
2)
3P(O)(OMe)
2 [41][48].
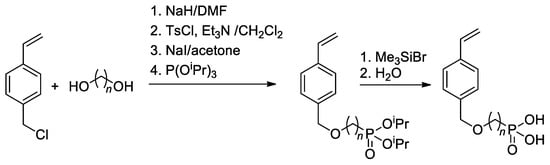
Since substituted styrenes are highly active in all types of polymerizations, the idea of using –P(O)(OH)
2-functionalized styrenes in polymerization is certainly feasible. Based on 4-(chloromethyl)-styrene, Yoon and Coll.
[42][49] have synthesized styryl phosphonic acids with different lengths of the alkanediyl spacers between the benzene ring and –P(O)(OH)
2 group (
Scheme 8). To introduce CH
2CF
2 linkage between styrene and phosphonate fragments, Alter and Hoge used the reaction of 4-(iodomethyl)styrene with LiCF
2P(O)(OEt)
2 [43][50].
Scheme 8. Synthesis of styrene-derivative monomers containing the phosphonic acid
[42][49].
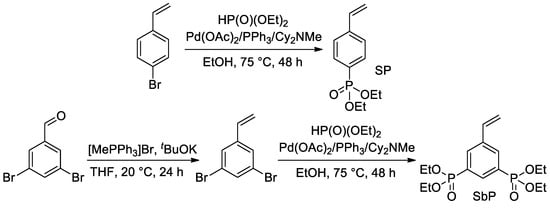
Later, Park et al. proposed the use of Pd-catalyzed cross-coupling for the direct introduction of one or two –P(O)(OEt)
2 fragments into an aromatic ring with a formation of styryl phosphonate and bis(phosphonate) (SP and SbP, respectively)
[44][51] (
Scheme 9).
Scheme 9. Synthesis of styrenes with one or two –P(O)(OEt)
2 substituents
[44][51].
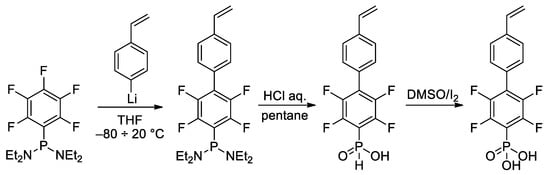
In the study of Hoge et al.
[45][52], the idea of the use of styrene-derived phosphonates was further developed. Considering the high
para-selectivity of nucleophilic substitution in N,N,N′,N′-tetraethyl-1-(perfluorophenyl)phosphinediamine, they synthesized vinyl monomer having 1,4-perfluorophenylene linkage between styryl and –P(O)(OH)
2 fragments (
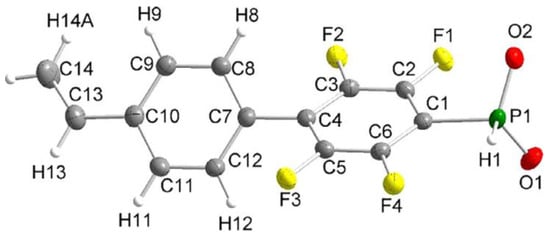
Scheme 10). Note that intermediate phosphinic acid is virtually the only phosphorus-containing ‘monomer’ with a proven molecular structure (
Figure 1).
Scheme 10. Synthesis of phosphonic acid with 1,4-perfluorophenylene linkage between styryl and –P(O)(OH)
2 fragments
[45][52].
Figure 1. Molecular structure of the anion of the diethyl ammonium salt of phosphinic acid with 1,4-perfluorophenylene linkage between styryl and –PO
2H
− fragments (thermal ellipsoids are set to 50% probability; cation omitted.). Reprinted with permission from
[45][52]. Copyright (2020) Wiley-VCH GmbH.
Dialkyl(alkenyl)phosphates, readily available by the reaction of enolates with ClP(O)(OR)
2 [46][47][53,54], can also be considered as monomers for the synthesis of sidechain PCPAs; however, their formation is complicated by the rearrangement to β-ketophosphonates
[48][49][55,56].
2.3. Homopolymerization and Copolymerization of the Phosphorus Containing Vinyl Monomers
2.3.1. Homopolymerization of VPA, Its Derivatives and Analogs
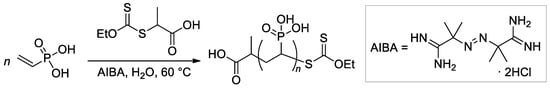
Since poly(vinylphosphonic acid) (PVPA) continues to attract the researchers’ attention based on new fields of PVPA can be synthesized via radical polymerization of vinylphosphonic acid in the presence of initiators and optionally chain transfer agents (CTAs) with different functionality and reactivity.
Wegner et al.
[50][57] conducted a series of experiments on VPA polymerization in aqueous media using α,α′-azodiisobutyramidine dihydrochloride (AIBA) as an initiator. In contrast to free-radical polymerization of other polar vinyl monomers, polymerization of VPA occurred with low regioselectivity, and a probability of head-to-head and tail-to-tail links over regular head-to-tail links of 23.5% was obtained. When using AIBN as an initiator and ethyl acetate as a solvent, PVPA containing up to 17% head-to-head fragments was obtained
[51][58]. To explain the low regioselectivity of VPA polymerization, Wegner et al.
[50][57] suggested that the process may occur according to two different mechanisms, i.e., a classical head-to-tail radical polymerization and a cyclopolymerization of the VPA anhydride formed in situ (
Scheme 11). Further studies on VPA polymerization in Ac
2O media, conducted by Voit and Coll.
[52][59], have shown that the intermediate formation of anhydride results in an acceleration of the reaction due to higher reactivity of the VPA anhydride with a formation of cyclic side-products (
Scheme 11). The formation of anhydride fragments (up to 19 wt%) was also detected when the polymerization of VPA was conducted in ethyl acetate with the use of a benzoyl peroxide initiator
[53][60]. AIBA-initiated homopolymerization of VPA was used in the preparation of PVPA samples suitable for composition with different inorganic compounds
[54][55][56][61,62,63].
Scheme 11. Formation of the side products during polymerization of VPA resulting from intermediate formation of the VPA anhydrides
[50][52][57,59].
The use CTAs in the radical polymerization of VPA was reported for the first time by David and Coll.
[57][64]. Only CBrCl
3 successfully served as CTA in the synthesis of telechelic VPA oligomers with yields of ~70% and
DPn = 6–60. High-MW PVPA (
Mn 42.2 kDa) was obtained in aqueous media at 80 °C when using a 620:1 VPA/AIBA ratio
[58][65].
Prospective results were received when using
n-C
6F
13I as a CTA that exhibited CF
2I functionalization of the oligomer obtained. MALDI-TOF MS and NMR analysis indicated that conventional radical initiation and termination do not occur and that the oligomers were obtained from transfer reactions
[59][66].
The applicability of reversible addition-fragmentation chain transfer/macromolecular design via the interchange of xanthate (RAFT/MADIX) approach to radical polymerization of VPA was studied by Destarac and Coll.
[60][67]. They demonstrated that at 65 °C in water carboxy functionalized O-ethyl xanthate reacts rapidly and quantitatively with VPA oligoradicals to yield low-MW O-ethyl xanthate-capped PVPAs (
Scheme 12), and later, reversible transfer of the xanthate terminal group leads to an increase of
Mn during polymerization. AIBA was used as a water-soluble initiator of radical polymerization. A follow-up study by the same group has shown that in the presence of 0.25-0.75, molar equivalents of NaOH, the rate and the final conversion of both conventional and RAFT polymerizations were increased, and the fastest rates of polymerization were obtained in the presence of 0.5 equivalents of NaOH
[61][68].
Scheme 12. Aqueous RAFT/MADIX polymerisation of VPA mediated by a carboxy-functional xanthate
[60][67].
The other possibility to obtain PVPA consists in the polymerization of vinylphosphonic acid derivatives CH
2=CHP(O)Cl
2 or CH
2=CHP(O)(OR)
2 followed by hydrolysis. As was demonstrated by Ellis and Wilson, PVPA can also be obtained by free-radical polymerization of CH
2=CHP(O)Cl
2 with the use of an AIBN initiator, followed by hydrolysis
[62][69]. However, to provide acceptable mechanical characteristics of PVPA-based materials, the relatively low-MW polymer thus obtained was forcedly subjected to the treatment with cross-linking reagents
[63][70].
Jin and Gonsalves reported a relatively low
Mn value of 8 kDa for homopolymer formed during AIBN-initiated bulk polymerization of CH
2=CHP(O)(OMe)
2 [64][71], the microstructure of the polymer was not determined. During the study of AIBA-initiated polymerization of CH
2=CHP(O)(OMe)
2, Wegner et al.
[50][57] showed higher regioselectivity of this reaction in comparison with VPA homopolymerization. For the transformation of the polymer obtained into PVPA, it reacted with an excess of aq. HBr at 110 °C for 8 h. Note that radical polymerization of CH
2=CHP(O)(O
iPr)
2 can be complicated by intramolecular hydrogen transfer from the backbone to the side chain
[65][72] (
Scheme 13).
Scheme 13. Intramolecular hydrogen transfer from the backbone to the side chain with formation of P–O–C bonds during radical polymerization of diisopropyl vinyl phosphonate (P = polymer)
[65][72].
The study of UV-induced copolymerization of VPA with CH
2=CHP(O)(OMe)
2 [66][73] deserves to be mentioned: in the presence of 3 wt% Darocur 4265 photoinitiator (that represents a mixture of (diphenylphosphoryl)(mesityl)methanone and 2-hydroxy-2-methyl-1-phenylpropan-1-one), VPA or VPA/phosphonate mixtures were laid on PTFE plates and exposed to UV irradiation. PVPA with
Mn = 27.75 kDa (
ÐM = 1.23) was obtained; for copolymers,
Mn values increased from 8.78 to 16.12 kDa with an increase in the VPA/CH
2=CHP(O)(OMe)
2 ratio from 1:1 to 4:1. ATRP was also used in the synthesis of core-shell materials with the use of oligo-(CMe
2Br) substituted polyaromatics and CH
2=CHP(O)(OEt)
2 [67][74].
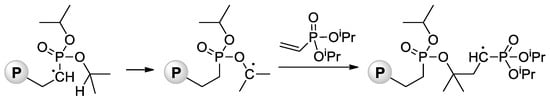
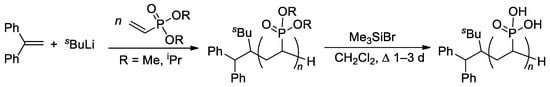
The relatively low reactivity of CH
2=CHP(O)(OR)
2 in free-radical polymerization has heavily conditioned the search for alternative methods of polymerization of dialkyl vinyl phosphonates. Meyer and Coll. proposed the use of
sBuLi and Ph
2C=CH
2 as an initiator of the anionic polymerization of CH
2=CHP(O)(OR)
2 (R = Me,
iPr) in THF
[68][75]. High yields of the polymers were obtained when using diisopropyl vinyl phosphonate, while CH
2=CHP(O)(OMe)
2 gave only low yields due to the precipitation of the polymer already in low conversions. Anionic polymerization proceeded regioselectively, but with low stereoselectivity. Given that mild and quantitative hydrolysis of dialkyl phosphonates can be performed via the intermediate formation of silyl esters
[69][76], polyesters were refluxed with Me
3SiBr in CH
2Cl
2 to yield PVPA samples of different
Mw (4.2–814 kDa) and
ĐM of 2.1–3.9 (
Scheme 14).
Scheme 14. Synthesis of PVPA by anionic polymerization of dialkyl vinyl phosphonate, followed by the cleavage using Me
3SiBr
[68][75].
A similar approach to PVPA via anionic polymerization of CH
2=CHP(O)(OMe)
2 with subsequent hydrolysis was implemented by Takeichi et al.
[70][77] with the use of
tBuLi or
tBuLi/
nBu
3Al (1:5 mol/mol) as an initiator at the first stage (polymerization was conducted in toluene media), and conc. HCl with 12 h of reflux at the second stage. When polymerization was initiated by
tBuLi/
nBu
3Al instead of
tBuLi alone, a threefold increase in monomer conversion was detected and, more significantly,
ÐM decreased by half, from 2.65 to 1.30, and isotacticity increased. The water solutions of the PVPAs were cast on a glass plate and allowed to stand at room temperature for seven days. After drying, the PVPA (
Mn = 5.0 kDa, m = 52%), prepared through the radical polymerization, became viscous liquids, whereas PVPA (
Mn = 5.5 kDa, m = 67%), prepared through the anionic process, formed self-standing transparent film. In this way, the quality of the PVPA obtained at –78 °C with the use of
tBuLi/
nBu
3Al initiator is
clearly—in the proper sense of the word—illustrated by
Figure 2.
Figure 2. Photograph of a PVPA film derived from PDMVP prepared with
tBuLi/
nBu
3Al at −78 °C. Reprinted with permission from
[70][77]. Copyright (2010) Wiley-VCH GmbH.
Both free-radical and anionic polymerization of CH
2=CHP(O)(OR)
2 are usually not able to provide high monomer conversions and desirable molecular weight characteristics and microstructure of the polymer. In polyolefin chemistry, a similar problem was effectively solved by the use of coordination catalysis. However, conventional Ziegler-Natta and single-site catalysts, suitable for α-olefin polymerization, cannot be applied in the coordination polymerization of phosphonates due to the high electron donation ability of the oxygen atom of →P=O fragment. To date, only rare earth metal complexes were used successfully to polymerize dialkyl vinyl phosphonates.
In 2010
[71][78], Rabe and Coll. reported that rare earth metal amides of the formula [N(SiHMe
2)
2]
3(THF)
2 (Ln = La, Nd, Sm) catalyze polymerization of CH
2=CHP(O)(OEt)
2 with a formation of high-MW isotactic polymers (
Mn = 65–84 kDa,
ÐM = 3.1–3.6, mm = 66–79%). A corresponding Y complex was found to be inactive in polymerization, forming a stable adduct [N(SiHMe
2)
2]
3[CH
2=CHP(O)(OEt)
2]
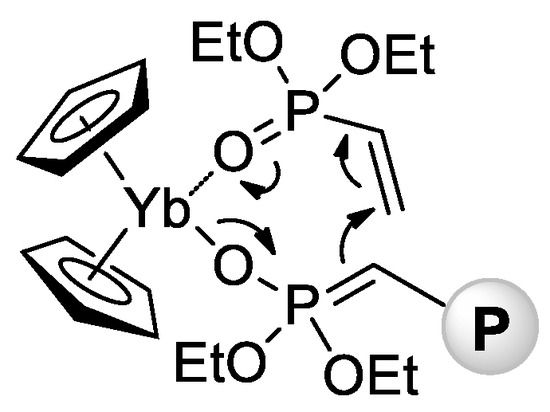
2. In the same year, Rieger and Coll. described the first example of metallocene-catalyzed polymerization of CH
2=CHP(O)(OEt)
2 [72][79]. The complexes (η
5-C
5H
5)
2YbX (Cp
2YbX, X = Cl, Me) have demonstrated high activities in toluene reaction media, polymerization had the ‘living’ character, and
Mw up to 1280 kDa were achieved. Already in this first work, Rieger et al. proposed a group-transfer mechanism of polymerization (
Scheme 15) that was confirmed and discussed in subsequent research articles
[73][74][75][76][77][78][79][80,81,82,83,84,85,86] and reviews
[80][81][87,88] of this scientific group.
Scheme 15. Postulated intermediate in the group-transfer mechanism of CH
2=CHP(O)(OEt)
2 polymerization, catalyzed by Cp
2YbX.
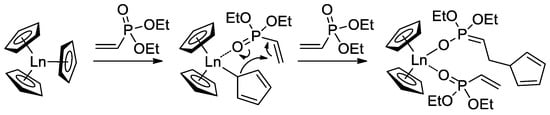
[72][79].
When using Cp
3Ln complexes (Ln = Lu, Yb, Tm, Er, Ho, Dy) as coordination catalysts of CH
2=CHP(O)(OEt)
2 polymerization, C
5H
5 fragment acted as an initiator of the chain growth (
Scheme 16)
[73][80]. Polymerization had the ‘living’ character, polymers with given
Mn (60–710 kDa depending on monomer-to-catalyst ratio) and
ÐM = 1.05–1.36 were obtained. Corresponding PVPAs were then prepared for the reaction with Me
3SiBr, followed by mild hydrolysis (HCl/MeOH). Dimethyl and diisopropyl vinyl phosphonates were also polymerized. DCS studies have shown that CH
2=CHP(O)(OR)
2-based polymers decompose at 280–340 °C (R = Et) and 245–270 °C (R =
iPr) with olefin elimination and formation of PVPA. When using Cp
3Yb catalyst, highly statistic copolymers of CH
2=CHP(O)(OR)
2 (R = Me, Et,
nPr) were obtained
[74][81].
Scheme 16. Initiation of the Cp
3Ln-induced polymerization of CH
2=CHP(O)(OEt)
2 [73][80].
Since Cp
2YbX-catalyzed polymerization of acrylates and CH
2=CHP(O)(OEt)
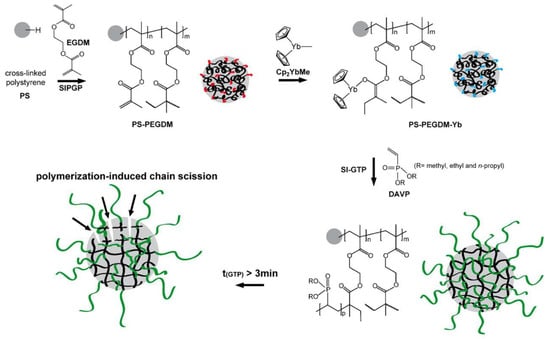
2 proceed according to similar group-transfer mechanisms, a surface-initiated group transfer polymerization (SI-GTP), which allows the covalent modification of solids with dense poly(vinylphosphonate) brushes, was carried out
[82][89]. Silicon surfaces were modified with methacrylate functionalities, either via a self-assembled monolayer of 3–(trimethoxysilyl)propyl methacrylate or via a poly(ethylene glycol dimethacrylate) film (prepared by self-initiated photografting and photopolymerization), the surfaces were treated with Cp
2YbMe, and then by vinyl phosphonate monomer. After Me
3SiBr and Me
3SiBr treatment, hydrophilic PVPA surfaces formed. A similar approach was realized using cross-linked polystyrene microspheres
[83][90] (
Figure 3).
Figure 3. Schematic illustration of the covalent grafting of a PEGDM on a cross-linked polystyrene microsphere and subsequent immobilization of the Cp
2YbMe. In the next step, SI-GTP of dialkyl vinyl phosphonate leads to the formation of poly(dialkyl vinyl phosphonate)-functionalized microspheres. Reprinted with permission from
[83][90]. Copyright (2014) Wiley-VCH GmbH.
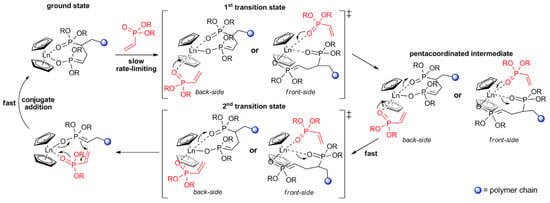
As a result of the mechanistic studies of polymerization of CH
2=CHP(O)(OR)
2 [75][82] it was demonstrated that initiation by Cp
2LnX follows a complex reaction pathway depending on the nature of X. For X = Me, CH
2SiMe
3 initiation occurs via the abstraction of the acidic α-CH of the vinylphosphonate, and for X = Cp, SR initiation occurs via the nucleophilic transfer of X to a coordinated monomer. For X = Cl, OR a monomer-induced ligand-exchange reaction with a formation of active Cp
3Ln catalyst was observed. The further elemental steps of the reaction are presented in
Figure 4 [75][82].
Figure 4. Elemental steps of rare earth-mediated group transfer polymerization of vinyl phosphonates. The rate-limiting step is an S
N2-type associative displacement of the polymer phosphonate ester by a vinylphosphonate monomer, presumably via a pentacoordinated intermediate. Reprinted with permission from
[75][82]. Copyright (2013) American Chemical Society.
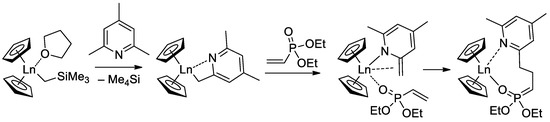
Since the initiation stage of Cp
2LnX-mediated polymerization of CH
2=CHP(O)(OR)
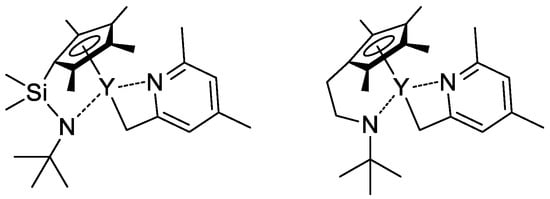
2, this stage is significantly slower than propagation stages (leading to the broadening of MWD and partial loss of the polymer chain control), subsequent studies were focused on the development of highly efficient initiators. In that capacity, Cp
2Ln derivatives of 2,4,6-trimethylpyridine were proposed
[76][83]. The first stage of the process is presented in
Scheme 17. Polymerization experiments have demonstrated the absence of the induction period when using this new initiator.
Scheme 17. Synthesis of Cp
2Ln(CH
2(C
5H
2Me
2N)) via C−H bond activation by σ-bond metathesis and initiation step of the CH
2=CHP(O)(OEt)
2 polymerization
[76][83].
The concept of the efficient initiator of rare earth metal-mediated group transfer polymerization was beautifully realized by the example of the core-first polymer synthesis
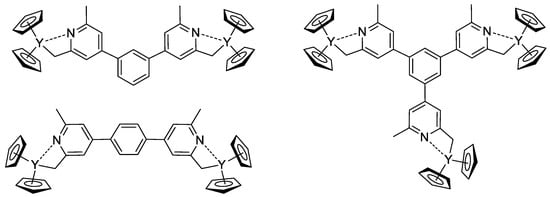
[78][85]. Based on 1,3- (or 1,4-bis-) and 1,3,5-tris(2,6-dimethylpyridin-4-yl)benzene, Cp
2Y complexes (
Scheme 18) were obtained and used as initiators of the group transfer polymerization of CH
2=CHP(O)(OEt)
2. When using dinuclear complexes, bimodal polymer distribution differing in molecular weight by a factor of two was detected. In the case of the trinuclear complex, trimodal molecular weight distribution was observed.
Scheme 18. Efficient initiators for two- and three-pronged polymerization of CH
2=CHP(O)(OEt)
2 [78][85].
Rieger and Coll. also drew attention to another important aspect of lanthanidocene-initiated polymerization of CH
2=CHP(O)(OR)
2, namely, steric factors (
Figure 5)
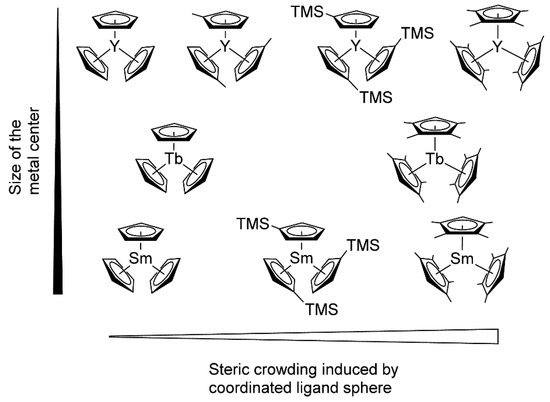
[77][84]. As a result of the metal−ligand interactions, the studied catalysts varied in their properties, ranging from inert (Cp
3Ln, Ln = Tb, Sm) to highly active ((C
5H
4Me)
3Y. (C
5Me
4H)
3Sm was the first example of the active ‘early’ lanthanidocenes, and in general, the activities accelerated with decreasing cation size. Furthermore, the polymerization behaviors of (C
5Me
4H)
3Ln complexes differed from unsubstituted or monosubstituted metallocenes. The pentacoordinate intermediates of these complexes exhibited lengthened metal-monomer bond lengths, resulting in a higher enthalpy ΔH
≠ term, and are partly compensated by lower ΔS
≠ contributions to the activation barrier.
Figure 5. Molecular structures of monosubstituted and tetramethyl-substituted Cp
3Ln catalysts studied in polymerization of CH
2=CHP(O)(OEt)
2. Reprinted with permission from
[77][84]. Copyright (2016) American Chemical Society.
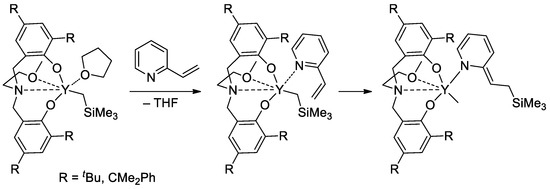
Rare-earth metal-based catalysts of the polymerization of vinyl phosphonates are not limited by sandwich lanthanide complexes. The high catalytic activity of the yttrium bis(phenolate) in the polymerization of CH
2=CHP(O)(OR)
2 (R = Et,
iPr) was demonstrated
[84][91], 2-vinylpyridine efficiently activated the pre-catalyst at the initiation stage (
Scheme 19).
Scheme 19.
Six-membered initiation mechanism for 2-vinylpyridine and Y chelate complexes in polymerization of CH
2
=CHP(O)(OR)
2
(R = Et,
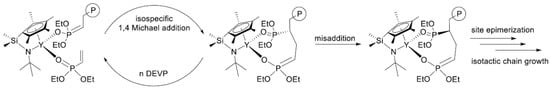
In their recent publication, Rieger and Coll. described the synthesis and catalytic behavior of half-sandwich ‘constrained geometry’ complexes of Y and Al in the polymerization of CH
2=CHP(O)(OEt)
2 [79][86]. The complexes of Y (
Scheme 20) have demonstrated high activity even at −78 °C, under these conditions highly isotactic polymer (
mmm > 98%) was obtained. The proposed stereocontrol mechanism is presented in
Figure 6.
Scheme 20. Efficient constrained-geometry catalysts for ‘living’ polymerization of vinyl phosphonates
[79][86].
Figure 6. Proposed stereocontrol mechanism and formation of stereoerrors for the isospecific polymerization of CH
2=CHP(O)(OEt)
2. Reprinted with permission from
[79][86]. Copyright (2019) American Chemical Society.
Note that in 2013
[85][92], Ni and Coll. have demonstrated high catalytic activity of Ln(BH
4)
3(THF)
3 (Ln = Y, La, Nd, Sm, Gd, Dy, Lu) in group transfer polymerization of CH
2=CHP(O)(OEt)
2. ‘Living’ character of the reaction was confirmed by the termination of the polymerization with the use of PhCH
2Cl or EtI, however, obtained polymers were characterized by broadened MWD (
ÐM = 1.63–1.81).
In conclusion, it is important to notice that the treatment with Me
3SiBr, followed by mild hydrolysis, or multi-hour reflux in HCl is not the only method for transforming poly(phosphonates) to PVPA. High efficiency of the use of CF
3SO
3H or highly acidic cationites in ‘wet’ (hydrolysis) and ‘dry’ (olefin elimination) transformation of dialkyl phosphonates to phosphonic acids was demonstrated by Han and Coll.
[86][93]. Different synthetic approaches to the hydrolysis of phosphinates and phosphonates have been reviewed very recently
[87][94], and nothing prevents researchers from using these methods in the synthesis of PVPA and other sidechain PCPAs.
2.3.2. Copolymerization of VPA and VPA Derivatives with Other Vinyl Monomers
Since VPA homopolymers have the prospective but limited potential of applications, valuable works on the synthesis of copolymers containing –CH
2CHP(O)(OH)
2– fragments were published even at the end of the last century.
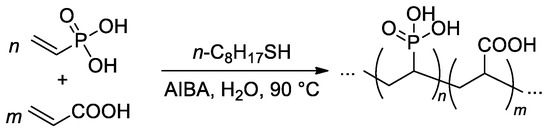
The free-radical copolymerization of acrylic acid and VPA was conducted by Budd and Coll. in aqueous media (
Scheme 21), AIBA was used as an initiator
[88][95]. As the VPA content in the feed was increased, the monomer conversion and yield of the copolymers showed a general decrease. The reactivity ratios of acrylic acid (r
1) and VPA (r
2) were 4.09 and 0.042, respectively. Such a high difference in r
1 and r
2 values may give rise to composition drift, where the composition of the copolymer changes as the polymerization proceeds. Where the VPA content in the feed was below 50%, a chain transfer agent was introduced into the polymerization to restrict the molecular weight. Using this method, a range of copolymer compositions were produced with consistent molecular weights (
Mw = 150–200 kDa) up to a VPA content of 59 mol%. At higher VPA contents, high-MW polymers were not obtained, and the highest
Mw for VPA homopolymer was 29 kDa.
Scheme 21. Free-radical copolymerization of acrylic acid and VPA
[88][95].
RAFT/MADIX approach was used in the synthesis of poly(acrylamide)-
b-PVPA copolymers (
Scheme 22)
[89][96] with a given length of poly(acrylamide) block and varied length of PVPA fragment, copolymers with
Mn 5.5–11.4 kDa were obtained.
Scheme 22. Aqueous RAFT/MADIX polymerization of VPA via chain extension of a polyacrylamide macroxanthate
[89][96].
When using ethylene glycol diacrylate, cross-linked copolymers of acrylic acid and VPA were obtained
[90][97]. Copolymers of VPA and 1-vinyl-1,2,4-triazole with 1:1, 1:2, and 2:1 comonomer ratios were obtained in DMF solution at 85 °C with AIBN initiator, the
Mn values ranged from 6.0 to 8.2 kDa, the dispersity
ÐM was ~2
[91][98]. Copolymerization of VPA and methacrylic ester of mPEG 475 was carried out in ethyl acetate with the use of AIBN as an initiator, side reactions and gel formation were detected
[92][99].
UV-initiated copolymerization of VPA and acrylamide at the oxidized silicon surface in the presence of 2,2-dimethoxy-2-phenylacetophenone (DMPA) was described in
[93][100], the binding of the copolymer with the surface was confirmed by photoelectron spectroscopy.
The synthesis of CH
2=CHP(O)(OMe)
2, CH
2=CF
2, and CH
2=CHSi(OEt)
3 terpolymers was described in
[94][101]. The best results were obtained when using 2,5-dimethyl-2,5-di(
tert-butylperoxy)hexane as an initiator of the reaction that was conducted in dimethyl carbonate at 115 °C. Si–O–Si cross-linking was accompanied by partial hydrolysis of methyl phosphonate fragments.
Poly(styrene)-
b-PVPA was obtained by
nBuLi-initiated sequential anionic polymerization of styrene and CH
2=CHP(O)(O
iPr)
2 in THF media with subsequent hydrolysis
[68][75].
The syntheses of VPA copolymers with 2-deoxy-2-methacrylamido-D-glucose, 4-acryloylmorpholine, or acrylamide, were the subject of the recent study of Nazarova and Coll.
[95][102]. The reactions were conducted in DMF or MeOH (AIBN initiator) or aqueous media (AIBA initiator). VPA has demonstrated values of reactivity comparable with 4-acryloylmorpholine and acrylamide, thus confirming the preference for the use of VPA instead of VPA esters in copolymerization.
With the use of (NH
4)
2S
2O
8 initiated free-radical polymerization in aqueous media, random terpolymers of VPA, acrylonitrile, and methyl acrylate were synthesized
[96][103]. Poly(vinylphosphonic acid-co-styrene-co-maleic anhydride) was obtained using AIBN as an initiator and DMSO as a solvent
[97][104]. Water-soluble initiator was used in the random copolymerization of VPA with 5-(methacryloylamido)tetrazole
[98][105].
As a final note, the use of rare-earth metal complexes in the synthesis of copolymers of vinyl phosphonates was the subject of the recent review of Rieger et al.
[81][88].
2.3.3. Homopolymerization and Copolymerization of Phosphorylated Acrylates
Derivatives of acrylic and methacrylic acid, containing –OP(O)(OH)
2 and –P(O)(OH)
2 fragments, have been evidently regarded as prospective monomers for the synthesis of sidechain PCPAs. There is extensive literature on this subject,
and resesarchersin this section, we have tried to present the most interesting and recent works in this field.
Evidently, one of the common methods of the polymerization of acrylates—anionic polymerization—is not applicable for derivatives of phosphoric and phosphinic acids, and the vast majority of research has used free-radical polymerization in one way or another. Trivial methods of free-radical polymerization (solution process, azo compounds, peroxides or UV exposure as initiators, no CTAs or other additives) are fully applicable to phosphorylated and phosphonated acrylates. This is particularly true in the case of acrylates, containing the phosphorus atom in an alkoxy fragment of the acrylate molecule (
Scheme 4), due to the high reactivity of these monomers, and a number of examples of the homo- and co-polymerization of phosphorylated and phosphonated (meth)acrylates can be cited
[15][16][18][19][21][23][24][30][31][32][99][100][101][22,23,25,26,28,30,31,37,38,39,106,107,108]. Note that the studies of AIBN-initiated copolymerization of methyl methacrylate and CH
2=C(Me)C(O)OCH
2P(O)(OMe)OH showed
r1 and
r2 values of 0.98 and 1.03, respectively, thus confirming close reactivity of ‘conventional’ and phosphonated acrylates
[23][30].
On the other hand, RAFT homo- and co-polymerization were successfully applied to similar acrylate monomers
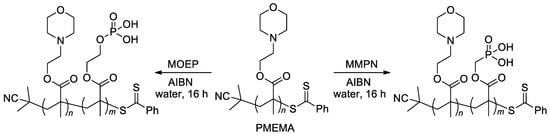
[22][26][102][29,33,109]. In particular, zwitterionic diblock copolymers were synthesized by copolymerization of the macro-RAFT agent, PMEMA (
Scheme 23) with methacrylate monomers MOEP and MMPN via RAFT polymerization
[22][29].
Scheme 23. Synthesis of diblock copolymers via RAFT polymerization
[22][29].
Copolymers of MPC and methyl acrylate with (4-formylphenyl) fragment via –(OCH
2CH
2)
9– spacer, were synthesized by RAFT polymerization
[103][110]. AIBN-initiated copolymerization of MPC,
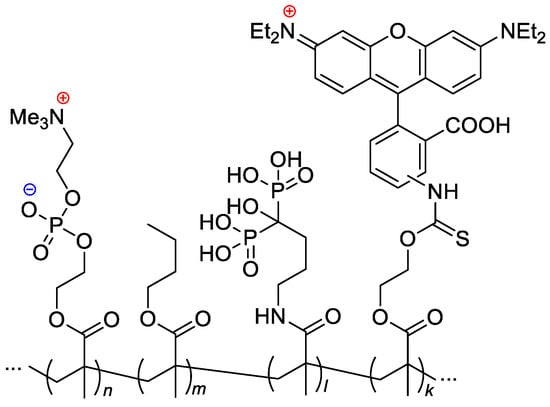
n-butyl methacrylate, MADP (optionally) and rhodamine-linked methacrylate (optionally, for the pharmacokinetic studies) resulted in copolymers with
Mn = 14–43 kDa and Ð
M ~2
[33][40] (
Scheme 24).
Scheme 24. Formulation of the bone-targeting phospholipid copolymers
[33][40].
The hyper-crosslinked polymer was obtained by AIBN-initiated free-radical polymerization of (CH
2=C(Me)C(O)OCH
2CH
2O)
2P(O)OH (BMEP) in DMF
[104][111].
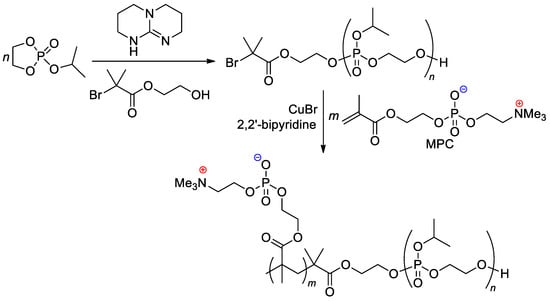
The zwitterionic block copolymer was synthesized by sequential carrying out of organocatalytic ROP of 2-isopropoxy-1,3,2-dioxaphospholane 2-oxide (
iPrOEP) with the use of BrCMe
2C(O)O(CH
2)
2OH as an initiator, and atom transfer radical polymerization (ATRP) of the phosphorylcholine-substituted methacrylate (MPC) in the presence of CuBr and 2,2′-bipyridine (
Scheme 25)
[105][112]. Note that MOEP-based polymer brushes were obtained with the use of the ATRP approach, based on an SH-substituted –CMe
2Br initiator bonded with gold nanoparticles
[106][113].
Scheme 25. Synthetic route to block copolymer containing both main-chain and sidechain phosphate fragments
[105][112].
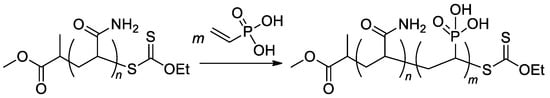
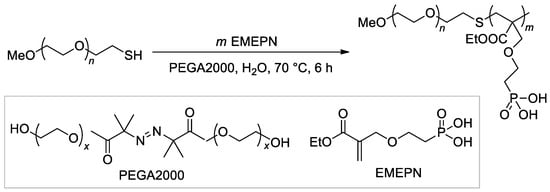
The synthesis of the diblock copolymer, containing mPEG-5000 fragment and polymer of (2-((2-(ethoxycarbonyl)allyl)oxy)ethyl)phosphonic acid as the second block
[107][114], is of particular interest due to the chosen method, ATRP with thiol CTA and macroinitiator PEGA2000 (
Scheme 26).
Scheme 26. The use of ATRP with PEGA2000 initiator in the synnthesis of hydrophilic diblock-copolymer
[107][114].
2.3.4. Homopolymerization and Copolymerization of Other Vinyl Monomers
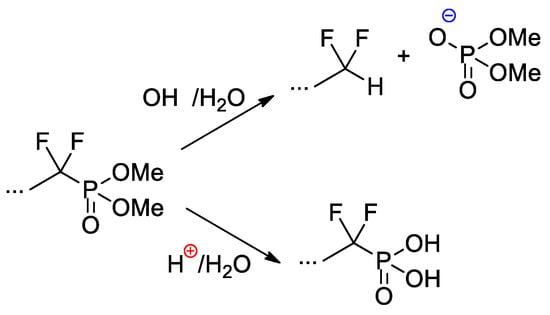
Copolymers of CF
2=CF
2, CF
2=CF(C
3F
7) and CF
2=CFO(CF
2)
3P(O)(OMe)
2 were obtained with the use of AIBN-initiated reaction in 1,1,2-trichloro-1,2,2-trifuoroethane (R-113) media
[41][48]. These copolymers as such have not demonstrated outstanding characteristics, but, nevertheless, a very significant chemical aspect should be mentioned here. Whereas hydrolysis of –P(O)(OR)
2-functionalized polymers is not accompanied by noteworthy side reactions, hydrolytic cleavage of the –CF
2P(O)(OMe)
2 fragment is not so straightforward (
Scheme 27). Under basic conditions, the cleavage of the C–P bond was observed, but acidic hydrolysis resulted in the formation of the phosphinic acid.
Scheme 27. Hydrolysis of perfluoroalkyl phosphonate fragment
[41][48].
For the synthesis of homopolymers of phosphonated styrenes SP and SbP (
Scheme 9) and their block copolymers with poly(isobutylene) Park and Coll. used ATRP in toluene media with CuCl/N,N,N′,N″,N″-pentamethyldiethylenetriamine catalytic system
[44][51]. Corresponding PCPAs were obtained by treatment with Me
3SiBr/CHCl
3 (36 h at 40 °C) followed by 8 h of methanolysis. Alter and Hoge
[43][50] have synthesized –CH
2CF
2P(O)(OH)
2-functionalized polystyrene (
Mn = 12.1 kDa,
ÐM = 2.42) by AIBN-initiated polymerization of styrene containing—CH
2CF
2P(O)(OEt)
2 substituent in the position 4, followed by two-stage deprotection (reaction with Me
3SiBr/CH
2Cl
2 and methanolysis).
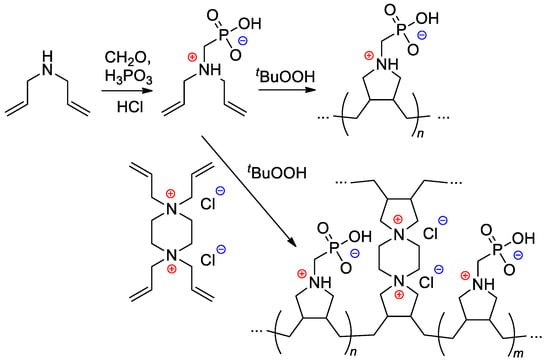
2.3.5. Cyclopolymerization
Starting from diallylamine, both linear and cross-linked PCPAs were synthesized by Al Hamouz and Ali
[108][115] by free-radical diene cyclopolymerization approach (
Scheme 28).
Scheme 28. Synthesis of phosphonate-containing diene monomer and its free-radical diene cyclopolymerization
[108][115].