Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Qun Zhou.
As hybrid molecules containing biologics and highly toxic low-molecular weight chemotherapeutic drugs, antibody–drug conjugates (ADCs) leverage the advantages of both targeting specificity of antibodies and high potency of cytotoxic compounds or synthetic cytotoxins. In addition to the use of synthetic cytotoxic compounds, many different payloads, including non-cytotoxic compounds, proteins/peptides, glycans, lipids, and nucleic acids, have been utilized for site-specific antibody conjugations.The site-specific antibody conjugates containing these payloads can be used in proof-of-concept studies or in developing new therapeutics for unmet medical needs.
- site-specific antibody conjugation
- engineering
- payloads
1. Introduction
Antibody–drug conjugation has gained significant momentum during the past few years with more than ten antibody–drug conjugates (ADCs) being approved by regulatory agencies for cancer treatment in clinics [1,2,3,4,5][1][2][3][4][5]. As hybrid molecules containing biologics and highly toxic low-molecular weight chemotherapeutic drugs, ADCs leverage the advantages of both targeting specificity of antibodies and high potency of cytotoxic compounds or synthetic cytotoxins. To synthesize ADCs, the antibodies are coupled with drug-linkers using different conjugation chemistries. The therapeutic index of the ADCs depends on many attributes including the expression profiles of selected cancer antigens, the qualities and specificities of antibodies, the properties of the synthetic cytotoxins (potency, mechanism of action, loading, cleavable or non-cleavable linkers), and the conjugation chemistries used [6]. The conventional conjugation approaches rely on non-specific/stochastic coupling of drug-linkers to lysines (about 40 residues per IgG1) or hinge cysteines (8 residues per IgG1). They often result in a heterogeneous profile of ADCs with a drug-to-antibody ratio (DAR) of 2 or 4, leading to difficulties in characterization and process control. To overcome these disadvantages, next generation site-specific antibody–drug conjugation methods have been developed. These methods have been reviewed in many excellent publications [7,8,9,10,11,12,13][7][8][9][10][11][12][13].
In addition to using synthetic cytotoxins, there is increased interest in coupling other payloads with site-specific antibody conjugation. These payloads include non-cytotoxic compounds that are not cytotoxic to human cells, as well as proteins/peptides, glycans, lipids, and nucleic acids.
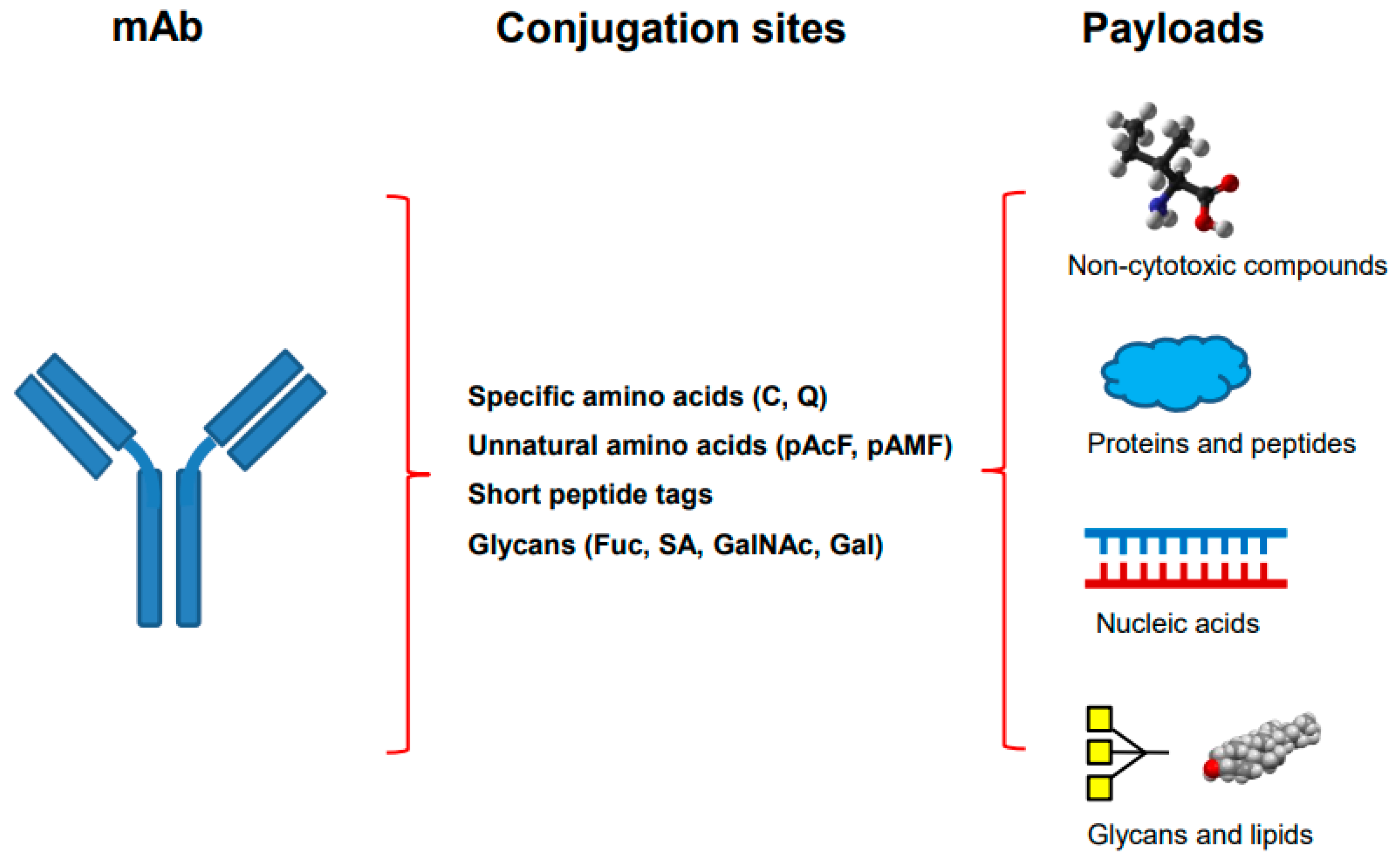
As described in many site-specific antibody conjugations, genetic engineering has been carried out by introducing specific sites for conjugation, such as cysteine (Cys), glutamine (Gln), unnatural amino acids (p-acetylphenylalanine or pAcF and p-azidomethyl-L-phenylalanine or pAMF), or short peptide tags. The mutated DNA is transfected into the cells to express engineered antibody. Cell line engineering is often required for the genetic engineering if the antibody of interest would be used for therapeutic developments. Once it is established, the conjugation process is straightforward. Metabolic labeling is applied for several methods, such as site-specific conjugation through unnatural amino acids, and its selectivity and efficiency have been demonstrated [24,25][24][25]. Since progress in site-specific antibody conjugations has been reviewed in detail previously [7[7][8][9][10][34][43][44],8,9,10,34,43,44], only the recent advances are highlighted here.
The conjugation through Cys is relatively simple and straightforward without the need for special reagents or enzymes, although there is potential instability in vivo, depending on location of the engineered Cys residue. THIOMABTM, the site-specific conjugation through engineered Cys, was one of the first site-specific antibody drug conjugation methods being developed [14]. The ADC generated using this method shows not only high homogeneity, but also increased efficacy and therapeutic index in vivo in animal models. There are many different sites in Fab and Fc regions that have been engineered to introduce single unpaired Cys residues for site-specific conjugation [15,45,46,47,48,49][15][45][46][47][48][49]. Recently, antibody engineering by introducing double and triple unpaired Cys has also been described leading to conjugates with more payloads (DAR of more than two) per antibody [22,50,51,52,53][22][50][51][52][53].
The unnatural amino acid can be introduced at different positions in an antibody providing the sites for stable conjugation. However, special reagents and extensive cell line engineering are often required for the approach.
Conjugations through chemoenzymatic modification methods have been demonstrated by using multiple enzymes including transglutaminase, transpeptidase sortases, glycosyltransferases or endoglycosidases. There is no need for genetic engineering with the conjugation through antibody glycans, but special reagents and enzymes are necessary. Enzymatic modifications of amino acids within specific short peptide tags generate antibody conjugates coupled with payloads at high selectivity and stability although the potential immunogenicity of introduced peptide tags is currently unknown. Although chemoenzymatic methods have been demonstrated as efficient processes, the reagents and associated cost may need to be considered for process scale-up. It was recently reported that the introduction of small-molecule drugs into Gln of an aglycosylated antibody by using microbial transglutaminase (mTG) can have an impact on the stability of ADC. The hydrophobic cytotoxin was able to compensate for thermal destabilization resulting from structural distortions due to antibody deglycosylation [21]. The site-specific conjugation of Q295 in deglycosylated antibody was also described by using the same transglutaminase with cystamine for thiolation [23]. The chemically introduced thiol on Q295 could be selectively conjugated using maleimide chemistry with improved plasma stability. In another report, Wijdeven et al. described an improved method from enzymatic glycan remodeling followed by metal-free click chemistry [35]. An engineered endoglycosidase and a native glycosyltransferase were selected for chemoenzymatic reaction using a novel azido sugar, generating ADCs with improved efficacy. Recently, there were advances from site-specific antibody conjugation using transglycosylation. A one-pot reaction for deglycosylation and transglycosylation in antibody Fc was described using wild-type endoglycosidase from Streptococcus pyogenes of serotype M49 (Endo-S2) [54,55][54][55]. The enzyme was shown to efficiently introduce the functionalized disaccharide oxazolines carrying site-selectively modified azide in varied numbers, resulting in ADCs with a precise control of DAR ranging from 2 to 12 via a copper-free strain-promoted click chemistry. Endo-S2 was able to accommodate drug-preloaded minimal disaccharide derivative oxazolines as donor substrates for efficient transfer of the glycan containing drug-linker. These ADCs containing monomethyl auristatin E (MMAE) with higher DARs were shown to be more potent in killing antigen-overexpressing cancer cells than those with lower DARs. The in vivo anticancer efficacy in tumor xenograft model was reported with MMAE-conjugated ADCs generated using a similar approach [56].
Among many different methods as described, the site-specific conjugations through engineered Cys, unnatural amino acid, and enzymatic glycan remodeling–metal-free click chemistry have been performed at big scale and the produced conjugates are being tested in clinical trials [11,35][11][35]. Besides ADCs, site-specific antibody conjugation methods have also been applied for coupling other payloads as described in following sections (Figure 1).
 Several PROTAC compounds have been identified for degradation of bromodomain-containing protein-4 (BRD4), a member of the bromodomain and extra-terminal (BET) family of protein, that functions as an epigenetic “reader” of acetylated histone lysine residues [66]. Although the lead degraders displayed extremely potent BRD4 degradation in vitro, several compounds exhibited unfavorable physiochemical characteristics and poor in vivo pharmacokinetic properties following intravenous or oral administration to mice. Therefore, the degrader was conjugated to an antibody against C-type lectin-like molecule-1 (CLL1 or CLEC12A), which was engineered with three unpaired cysteine residues (LC-K149C, HC-L174C, and HC-Y373C), through methanethiosulfonyl-Cys conjugation. Despite the relatively high lipophilicity of the PROTAC-linker, it was still possible to attach six degraders per antibody (drug-antibody ratio or DAR of ~6). A single intravenous administration of the antibody–degrader conjugate led to dose-dependent tumor growth inhibition in mice bearing HL-60 (acute myeloid leukemia) cells xenografts. In another study, other BRD4 degraders were conjugated by the same laboratory to an antibody against six transmembrane epithelial antigen of the prostate 1 (STEAP1) overexpressed in prostate cancer [51]. The antibody was introduced with either a single Cys (LC-K149C) or three unpaired Cys (LC-K149C, HC-L174C, and HC-Y373C) and conjugated with BRD4 degraders with DAR of ~2 and ~6, respectively, using thiol-maleimide chemistry. The conjugates exhibited intracellular delivery of the payloads to PC3-S1 prostate cancer cells along with reductions in intracellular BRD4 levels and in MYC transcription in vitro. The same laboratory identified additional potent BRD4 degraders and converted them to protease-cleavable linkers bearing methanethiosulfonate functionality [52]. These degrader-linkers were coupled to the antibody against STEAP1 engineered with three unpaired Cys as described above with DAR of close to 6. The conjugates exhibited highly potent BRD4 degradation and antiproliferation activity against prostate cancer cell line PC3-S1 in vitro. The antibody conjugates bearing BRD4 degraders also showed strong anticancer efficacy in vivo in mouse xenograft assessments that employ several different cancer models. For selective protein degradation in breast cancer cells, a BRD4 degrader was conjugated to anti-HER2 (trastuzumab) by a different laboratory [65]. The antibody-PROTAC conjugate was prepared by using a novel dibromomaleimidestrained alkyne linker to rebridge the partially reduced interchain disulfide bonds and then coupling the protein degrader using copper-free strain-promoted azide–alkyne cycloaddition. The conjugate was internalized, resulting in active PROTAC for BRD4 degradation only in HER2 positive breast cancer cell lines in vitro. Furthermore, the field on selective degradation of estrogen receptor has also been advanced [75]. The site-specific antibody conjugation was described by using degraders that target estrogen receptor alpha (ERα) [50,77][50][77]. Anti-HER2–PROTAC conjugates containing ERα degraders were prepared using the same methods as described above by coupling selectively to engineered Cys residues [50]. They showed reasonably favorable in vivo stability and the degrader payloads were efficiently released after internalization in vitro.
The site-specific antibody conjugation with PROTAC provides an alternative approach to administer chimeric degrader compounds in vivo or to deliver them into specific cells or tissues. It may be very useful for PROTACs with sub-optimal drug properties and pharmacokinetics or need in cell/tissue specific delivery.
Several PROTAC compounds have been identified for degradation of bromodomain-containing protein-4 (BRD4), a member of the bromodomain and extra-terminal (BET) family of protein, that functions as an epigenetic “reader” of acetylated histone lysine residues [66]. Although the lead degraders displayed extremely potent BRD4 degradation in vitro, several compounds exhibited unfavorable physiochemical characteristics and poor in vivo pharmacokinetic properties following intravenous or oral administration to mice. Therefore, the degrader was conjugated to an antibody against C-type lectin-like molecule-1 (CLL1 or CLEC12A), which was engineered with three unpaired cysteine residues (LC-K149C, HC-L174C, and HC-Y373C), through methanethiosulfonyl-Cys conjugation. Despite the relatively high lipophilicity of the PROTAC-linker, it was still possible to attach six degraders per antibody (drug-antibody ratio or DAR of ~6). A single intravenous administration of the antibody–degrader conjugate led to dose-dependent tumor growth inhibition in mice bearing HL-60 (acute myeloid leukemia) cells xenografts. In another study, other BRD4 degraders were conjugated by the same laboratory to an antibody against six transmembrane epithelial antigen of the prostate 1 (STEAP1) overexpressed in prostate cancer [51]. The antibody was introduced with either a single Cys (LC-K149C) or three unpaired Cys (LC-K149C, HC-L174C, and HC-Y373C) and conjugated with BRD4 degraders with DAR of ~2 and ~6, respectively, using thiol-maleimide chemistry. The conjugates exhibited intracellular delivery of the payloads to PC3-S1 prostate cancer cells along with reductions in intracellular BRD4 levels and in MYC transcription in vitro. The same laboratory identified additional potent BRD4 degraders and converted them to protease-cleavable linkers bearing methanethiosulfonate functionality [52]. These degrader-linkers were coupled to the antibody against STEAP1 engineered with three unpaired Cys as described above with DAR of close to 6. The conjugates exhibited highly potent BRD4 degradation and antiproliferation activity against prostate cancer cell line PC3-S1 in vitro. The antibody conjugates bearing BRD4 degraders also showed strong anticancer efficacy in vivo in mouse xenograft assessments that employ several different cancer models. For selective protein degradation in breast cancer cells, a BRD4 degrader was conjugated to anti-HER2 (trastuzumab) by a different laboratory [65]. The antibody-PROTAC conjugate was prepared by using a novel dibromomaleimidestrained alkyne linker to rebridge the partially reduced interchain disulfide bonds and then coupling the protein degrader using copper-free strain-promoted azide–alkyne cycloaddition. The conjugate was internalized, resulting in active PROTAC for BRD4 degradation only in HER2 positive breast cancer cell lines in vitro. Furthermore, the field on selective degradation of estrogen receptor has also been advanced [75]. The site-specific antibody conjugation was described by using degraders that target estrogen receptor alpha (ERα) [50,77][50][77]. Anti-HER2–PROTAC conjugates containing ERα degraders were prepared using the same methods as described above by coupling selectively to engineered Cys residues [50]. They showed reasonably favorable in vivo stability and the degrader payloads were efficiently released after internalization in vitro.
The site-specific antibody conjugation with PROTAC provides an alternative approach to administer chimeric degrader compounds in vivo or to deliver them into specific cells or tissues. It may be very useful for PROTACs with sub-optimal drug properties and pharmacokinetics or need in cell/tissue specific delivery.
2. Overview of Site-Specific Antibody Conjugation
Site-specific antibody conjugation begins with the engineering or modification of monoclonal antibody, followed by the conjugation of optimized drug-linkers (Figure 1). The antibody is engineered through the Fab or Fc region of an IgG to introduce different conjugation sites by using genetic engineering, metabolic labeling or chemoenzymatic modification. Many different methods can be categorized through different conjugation sites in the antibody (Table 1, Figure 1).Figure 1. The site-specific antibody conjugation using payloads other than synthetic cytotoxins. The monoclonal antibody on the left is engineered by introducing different sites for selective coupling including specific amino acids, unnatural amino acids, short peptide tags, or modified glycans (middle). Different payloads, including non-cytotoxic compounds, proteins and peptides, nucleic acids, as well as glycans and lipids (right), are used for conjugation.
Table 1.
The four categories of the site-specific antibody–drug conjugation.
| Techniques | Conjugation Sites | Genetic Engineering | Metabolic Labeling | Chemo-Enzymatic Modification | Selective References |
|---|---|---|---|---|---|
| Specific amino acids | C (Cys), Q (Gln) | + | − | ± | [14,15,16,17,18,19,20,21,22,23][14][15][16][17][18][19][20][21][22][23] |
| Unnatural amino acids | pAcF, pAMF, Sec, etc. | + | + | ± | [24,25,26,27,28][24][25][26][27][28] |
| Glycans | Sialic acid, GalNAc, GlcNAc, Gal, Fuc, etc. | − | ± | + | [29,30,31,32,33,34,35][29][30][31][32][33][34][35] |
| Short peptide tags | LLQG, LCTPSR, etc. | + | − | + | [36,37,38,[39,40,3641,42]][37][38][39][40][41][42] |
3. Non-Cytotoxic Compounds as Payloads
There are several non-cytotoxic compounds used for selective conjugation (Table 2). They include polyethylene glycol (PEG), antibiotics, immuno-modulating compounds, protein degraders, and ligands for receptors and proteins that are overexpressed in cancer.Table 2.
Site-specific antibody conjugations with non-cytotoxic compounds.
| Categories of Payloads | MOA of Payload | Antibody Formats | Specific Site Used | Conjugation Chemistry | References |
|---|---|---|---|---|---|
| PEG | Prolonged serum half-life | Fab | Engineered Cys in C-terminus | Thiol-mediated conjugation | [57,58,59][57][58][59] |
| Antibiotics | Inhibitor of bacterial RNA polymerase | mAb | LC V205C | THIOMABTM | [60,61][60][61] |
| Immune-modulating compounds | PDE4 inhibitor for immune suppression | mAb | Unnatural amino acid | Oxime chemistry | [62] |
| Liver LXR agonist for immune suppression | mAb | Unnatural amino acid | Oxime chemistry | [63] | |
| Agonist of glucocorticoid receptor | Nb | C-terminal LPETGG | Sortase A (SrtA) mediated transpeptidation | [64] | |
| Protein degraders | PROTAC-mediated ERa and BRD4 degradation | mAb | Engineered Cys | THIOMABTM | [50] |
| PROTAC-mediated BRD4 degradation | mAb | Hinge Cys | Click chemistry | [65] | |
| mAb | Engineered Cys | THIOMABTM | [51,52,66][51][52][66] | ||
| LYTAC-mediated degradation through ASGPR | mAb | FGly in C-terminus of HC, hinge, CH1 | Hydrazino-iso-Pictet–Spangler reaction and click chemistry | [67] | |
| LYTAC-mediated degradation through M6PR | mAb | N-glycans | Chemoenzymatic reaction | [68] | |
| Ligand for proteins overexpressed in cancer | Chemically programmed bispecific Fab as T-cell engager (Fab-synthetic ligands) | Fab | Unnatural amino acid at HC K138 | Oxime chemistry | [69] |
| Fab | C-terminal Sec in HC | SeH-maleimide chemistry | [70] |
3.1. PEG
To prolong the serum half-life of antibody fragments, the PEGylation of Fab through hinge Cys residues was first described more than 20 years ago [71]. Later research on Fab engineering led to PEGylation at introduced unpaired Cys residue at the C-terminal end of the heavy chain (HC) constant region 1 [57,58,59][57][58][59]. Since the attachment of PEG (20 to 40 kDa in size) is far away from the epitope binding region, the antigen binding and in vitro bioactivity of PEGylated Fab are usually not reduced as compared to unmodified Fab. The PEGylation also does not affect stability, while the half-life of the antibody fragment can be significantly increased. A PEGylated anti-TNF Fab, Certolizumab pegol, was approved by regulatory agencies for treating patients with immune-mediated inflammatory diseases, while other PEGylated Fab constructs are under proclinical and clinical developments. It would be interesting to compare pharmacokinetics and pharmacodynamics of PEGylated Fab, which is monovalent and has no effector function, with the bivalent antibody IgG.3.2. Antibiotics
A novel antibody–antibiotic conjugate (AAC) was described as a potential therapeutic that effectively kills intracellular bacteria [60]. The anti-S. aureus antibody was cloned and purified from B cells derived from the peripheral blood of patients recovering from various S. aureus infections. It was selected against wall-teichoic acids, pathogen-specific polyanionic glycopolymers that are connected to the thick peptidoglycan layers of gram-positive bacteria. A highly efficacious antibiotics rifalogue, which is activated only after being released in lysosomes, was conjugated to the antibody engineered with V205C at the light chain (LC) using the THIOMABTM approach. The AAC is superior to vancomycin for treatment of bacteremia in vivo and elimination of intracellular S. aureus infections. It substantially reduced bacterial load in the heart, kidney, and bones from mice on 7 and 14 days after a single intravenous administration [61]. The AAC provides a unique therapeutic approach against intracellular bacterial infection, and it is currently under clinical developments (ClinicalTrials.gov Identifier: NCT03162250).3.3. Immune-Modulating Compounds
Synthetic non-cytotoxic enzyme inhibitors or ligands for specific receptors have been coupled to antibodies through site-specific conjugations for immune modulation. An enzyme inhibitor to phosphodiesterase 4 (PDE4) has been utilized as a payload [62]. Although many PDE4 inhibitors have demonstrated anti-inflammatory activities, some of them showed dose-limiting side effects. To exploit tissue-restricted delivery of these inhibitors for increasing therapeutic index, a highly potent PDE4 inhibitor was coupled to an anti-CD11a, a surface antigen highly expressed by leukocytes including myeloid cells and lymphocytes. The antibody, which was engineered with the unnatural amino acid, pAcF, at A122 of the heavy chain as well as other mutations to silence effector functions, was selectively conjugated with aminooxy containing inhibitor using oxime chemistry. The immunoconjugate was rapidly internalized into immune cells and suppressed lipopolysaccharide-induced TNFα secretion in primary human monocytes. It displayed an in vivo anti-inflammatory effect in mouse models. The myeloid-cell-specific anti-Ly6C/G VHH molecules were also conjugated with dexamethasone, which contains an acid-labile hydrazone moiety, using sortase A (SrtA) mediated transpeptidation [64]. The conjugates enabled specific delivery of the dexamethasone, which has undesirable side effects, onto bronchial epithelium in influenza virus-infected mice. The VHH conjugates, but not free dexamethasone, reduced the weight loss of animals infected with virus. Several liver X receptor (LXR) agonists were investigated as a potential therapy for diseases including atherosclerosis based on their ability to induce reverse cholesterol transport and anti-inflammation [63]. However, they induced excessive lipogenesis in liver through their interaction with LXR-α. To prevent the on-target adverse effect, the LXR agonist was coupled to anti-CD11a, which was selected for targeting since the protein is highly expressed in macrophage and monocytes but not hepatocytes. The increased expression of CD11a on monocytes has been found to be correlated with atherosclerotic coronary stenosis [72]. An unnatural amino acid, pAcF, was introduced at position A122 of the antibody’s HC and conjugated with an aminooxy containing LXR agonist with a cleavable linker sensitive to lysosomal cathepsin B. The immunoconjugate induced LXR activation specifically in human THP-1 monocyte and macrophage cells in vitro, while it had no significant effect in hepatocytes. It would be interesting to see more research with antibody conjugated with immune modulating compounds being tested in different disease models.3.4. Protein Degraders: PROTAC
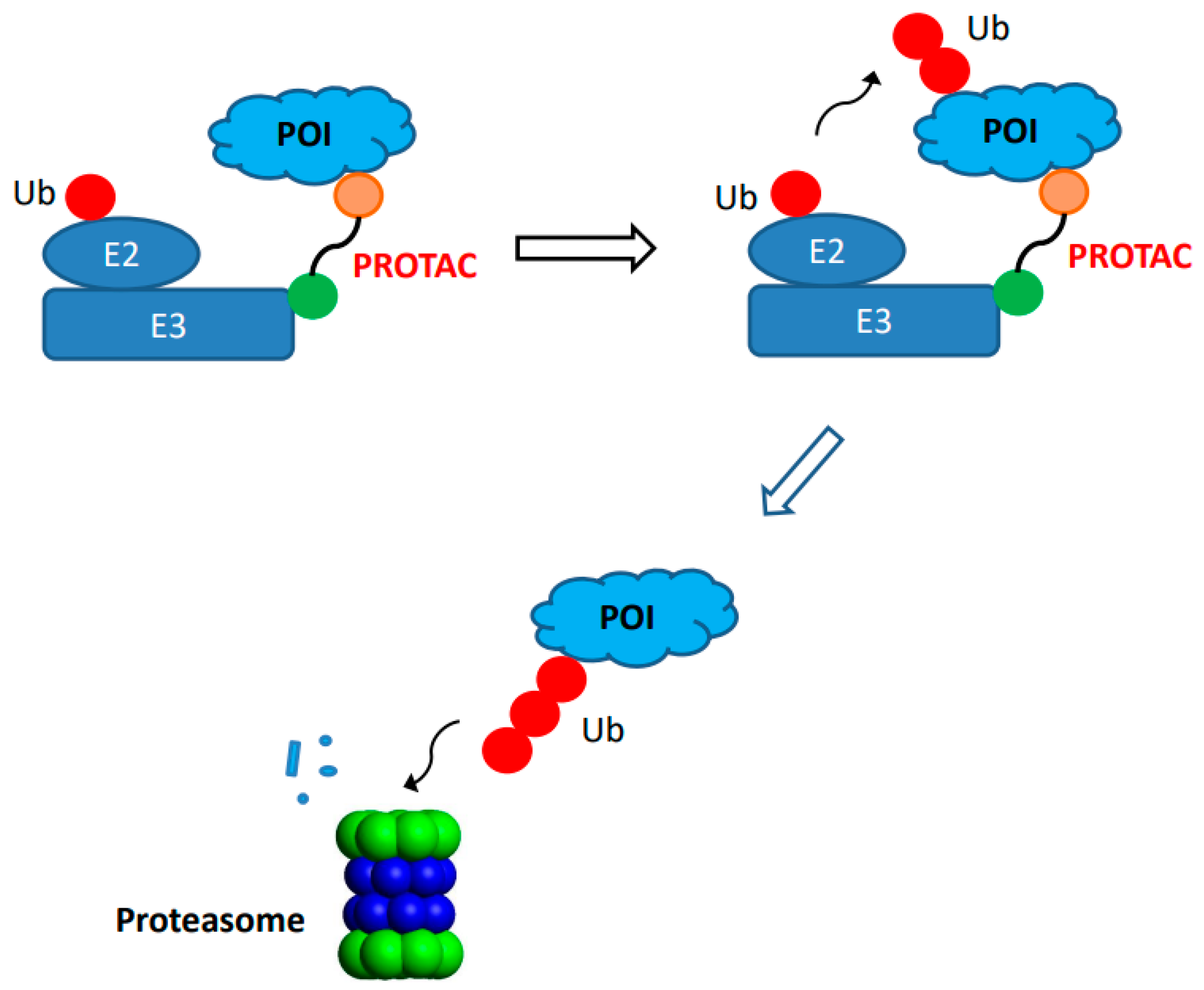
Recently, the site-specific antibody conjugation was applied to the field of protein degradation. The concept of proteolysis-targeting chimera (PROTAC) was first introduced more than 20 years ago [73,74][73][74]. It relies on synthetic chimeric molecule for intracellular degradation of protein target or protein of interest (POI) through the proteasome (Figure 2). The PROTAC degraders often contain three components: a ligand that binds to the POI, a ligand for E3 ubiquitin ligase, and a space group or linker that connects the first two components. During action, the PROTAC degrader, except for molecular glues, forms a ternary complex among itself, a POI, and an E3 ubiquitin ligase that leads to ubiquitination of the target for subsequent destruction via trafficking to the proteasome. The PROTAC technology makes it feasible for effective degradation of many intracellular protein targets, such as tyrosine kinases, hormone receptors, and transcription factors which are often undruggable via many conventional inhibitors. It also provides different mechanism of actions (event-driven pharmacology) as compared to conventional inhibitors (occupancy-driven pharmacology) [75]. Its potential to expand the application in drug discovery has generated great interests, resulting in significant progress during the last decade with several PROTAC degraders reaching clinical trials [75,76][75][76]. However, there are still some challenges associated with physico-chemical properties of some degraders such as relatively large entities that may compromise oral bioavailability, solubility, and/or in vivo pharmacokinetics [53]. To provide an alternate approach to administering chimeric degrader compounds in vivo, significant efforts have been made to generate antibody-PROTAC conjugates using site-specific conjugation approaches.Figure 2. Target protein degradation through proteolysis-targeting chimeras (PROTAC). A bifunctional small molecule PROTAC compound is consist of ligand (circle in orange color) that binds the protein of interest (POI, light blue) and ligand (circle in green) for an E3 ubiquitin ligase (E3, rectangle in dark blue). Both ligands are connected by a linker. The binding of PROTAC compound to both POI and E3 ubiquitin ligase results in ternary complex formation, leading to addition of ubiquitin (circle in red) to POI that is degraded by proteasome inside the cells.
3.5. Protein Degraders: LYTAC
In addition to PROTAC, site-specific conjugation has also been applied for lysosome-targeting chimeras (LYTAC) [78]. LYTAC relies on the interaction of ligands with its lysosomal-targeting receptor driving the degradation of extracellular protein targets or proteins of interest (POI), into the lysosome for degradation. It contains two parts: one is the ligand for lysosomal-targeting receptor while the other is the antibody against extracellular POI. Banik et al. prepared LYTAC by performing non-specific conjugation of antibodies against multiple extracellular POI with mannose-6-phosphonate polymer that binds to cation-independent mannose-6-phosphate receptor (CI-MPR) [79]. They showed efficient degradation of soluble proteins (APOE4) and membrane proteins (EGFR, CD71 and PD-L1) in cell lines in vitro. Recently, site-specific antibody conjugation was reported for the generation of LYTAC using chemoenzymatic methods [68]. Antibodies against HER2 (trastuzumab) and EGFR (cetuximab) were modified using endoglycosidase S for the deglycosylation and transfer of a synthetic high-affinity mannose-6-phosphate (M6P) glycan oxazoline, generating antibody-M6P glycan conjugates. The M6P containing LYTACs were able to selectively degrade membrane HER2 and EGFR in vitro. In addition to the CI-MPR, the degradation of POI with LYTAC binding to another lysosomal-target receptor, the asialoglycoprotein receptor (ASGPR), was also demonstrated [67,80][67][80]. Ahn et al. developed LYTACs that engage ASGPR, which is only expressed in hepatocytes, to degrade extracellular soluble and membrane proteins [67]. Antibodies against extracellular protein targets were conjugated with a triantenerrary N-acetylgalactosamine (GalNAc) that binds ASGPR in high affinity. A reactive aldehyde handle from formylglycine (FGly) was generated by FGly generating enzyme (FGE)-catalyzed oxidation of a specific Cys residue from an introduced short peptide tag, LCTPSR, in the antibodies. It was then conjugated to triantenerrary GalNAc using a hydrazine-iso-Pictet–Spangler reaction and copper-free click chemistry [81]. The antibody-GalNAc conjugate displayed efficient degradation of membrane receptors EGFR and HER2 on hepatocellular carcinoma cells, such as HepG2 cells, in vitro. The work has provided an example of targeted protein degradation in specific tissue such as liver.3.6. Ligand for Proteins Overexpressed in Cancer
There are reports on site-specific conjugation of antibodies with ligands for proteins overexpressed in cancers [69,70][69][70]. The anti-CD3 Fab or single-chain variable fragment antibody (scFv) was coupled to these ligands for the generation of bispecific T-cell engagers. These bispecific T-cell engagers are unique in that an anti-CD3 Fab or scFv is coupled with only a synthetic compound as ligands but is not linked to another antibody against cancer-associated proteins. In one study, an unnatural amino acid, pAcF, was incorporated into two different locations (LC-S202 and HC-K138) of the anti-CD3 Fab that are distal to antibody paratope based on its crystal structure [69]. The engineered Fab was conjugated to a synthetic high-affinity ligand for prostate-specific membrane antigen (PSMA). The bispecific T-cell engager showed potent cytotoxicity against prostate cancer cell lines in vitro and strong anticancer efficacy in vivo. In another study, high-affinity ligands for folate receptor α and integrin α4β1, which are overexpressed in multiple cancer cells, were coupled into introduced C-terminal selenocysteine (Sec) of an engineered anti-CD3 Fab [70]. The bispecific antibody–synthetic compound conjugates displayed potent cytotoxicity in vitro and ex vivo against cancer cell lines and primary cancer cells in the presence of T cells. They may be useful for screening or designing better biologics if the synthetic ligands are available. Chemically programmed antibodies (cpAbs) have been described for almost twenty years [69,82,83,84][69][82][83][84]. They were generated by using site-specific and covalent conjugation of small molecules to non-targeting mAbs with unique reactivity centers, such as an antibody against 1,3-diketone that was generated using reactive immunization. A unique reactive (usually nucleophilic) residue, such as lysine, in the antibody was coupled to synthetic compounds or peptides with a reactive (usually electrophilic) group. In the case of the anti-1,3-diketone antibody, the reversible covalent interaction of the lysine residue in the reactivity center with the 1,3-diketone forms an enaminone stabilized by an imine-enamine tautomerism. The cpAbs rely on coupled synthetic compounds for the recognition of extracellular antigens. Walseng et al. reported how a diabody containing both anti-hapten and anti-CD3 Fv (disulfide-linked polypeptides containing either VH or VL) can be coupled with hapten-derivatized folate through a reactive lysine introduced in one of the polypeptides of anti-hapten antibody [85]. The chemically programmed diabody demonstrated high selectivity and potency against folate receptor α-expressing ovarian cancer cells both in vitro and in vivo. The work is very interesting, but the potential immunogenicity of those constructs in human is unknown. Antibodies against HIV gp41 were engineered for site-specific conjugation with cholesterol [86]. The unpaired cysteine residues were introduced into antibodies as T20C on VL and S444C on HC for conjugation with maleimide containing cholesterol. It was demonstrated that the antibody–cholesterol conjugate could rescue antiviral activity of a mutant of a broadly neutralizing anti-HIV antibody with hydrophobic CDR H3 loops. The cholesterol component provided enrichment of the conjugate in lipid raft of the plasma membrane, facilitating recognition of protein epitope from the membrane-proximal external region of HIV gp41. The antibody conjugate also increased the antiviral activity of that wild-type antibody as well as another non-membrane-binding HIV antibody. Antibody conjugates containing non-cytotoxic compounds, which have been generated using site-specific conjugation, demonstrated impressive in vitro and in vivo results. It may be interesting to compare them with other antibody formats, such as those being generated using recombinant approaches, if it is feasible, in proof-of-concept studies. Nevertheless, they have shown potential as unique therapeutics for unmet medical needs.References
- Lucas, A.T.; Moody, A.; Schorzman, A.N.; Zamboni, W.C. Importance and Considerations of Antibody Engineering in Antibody-Drug Conjugates Development from a Clinical Pharmacologist’s Perspective. Antibodies 2021, 10, 30.
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the ‘high-hanging fruit’. Nat. Rev. Drug Discov. 2018, 17, 197–223.
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847.
- Dean, A.Q.; Luo, S.; Twomey, J.D.; Zhang, B. Targeting cancer with antibody-drug conjugates: Promises and challenges. mAbs 2021, 13, 1951427.
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39.
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804.
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337.
- Zhou, Q. Site-Specific Antibody Conjugation for ADC and Beyond. Biomedicines 2017, 5, 64.
- Zhou, Q.; Kim, J. Advances in the Development of Site-Specific Antibody-Drug Conjugation. Anti-Cancer Agents Med. Chem. 2015, 15, 828–836.
- Hussain, A.F.; Grimm, A.; Sheng, W.; Zhang, C.; Al-Rawe, M.; Bräutigam, K.; Abu Mraheil, M. Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches. Pharmaceuticals 2021, 14, 343.
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353.
- Schneider, H.; Deweid, L.; Avrutina, O.; Kolmar, H. Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal. Biochem. 2020, 595, 113615.
- Krüger, T.; Dierks, T.; Sewald, N. Formylglycine-generating enzymes for site-specific bioconjugation. Biol. Chem. 2019, 400, 289–297.
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932.
- Jeffrey, S.C.; Burke, P.J.; Lyon, R.P.; Meyer, D.W.; Sussman, D.; Anderson, M.; Hunter, J.H.; Leiske, C.I.; Miyamoto, J.B.; Nicholas, N.D.; et al. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconj. Chem. 2013, 24, 1256–1263.
- Dimasi, N.; Fleming, R.; Zhong, H.; Bezabeh, B.; Kinneer, K.; Christie, R.J.; Fazenbaker, C.; Wu, H.; Gao, C. Efficient Preparation of Site-Specific Antibody-Drug Conjugates Using Cysteine Insertion. Mol. Pharm. 2017, 14, 1501–1516.
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, J.; Tommasi, R.; Henseleit, K.; et al. In Vitro and In Vivo Evaluation of Cysteine Rebridged Trastuzumab-MMAE Antibody Drug Conjugates with Defined Drug-to-Antibody Ratios. Mol. Pharm. 2015, 12, 1872–1879.
- Behrens, C.R.; Ha, E.H.; Chinn, L.L.; Bowers, S.; Probst, G.; Fitch-Bruhns, M.; Monteon, J.; Valdiosera, A.; Bermudez, A.; Liao-Chan, S.; et al. Antibody-Drug Conjugates (ADCs) Derived from Interchain Cysteine Cross-Linking Demonstrate Improved Homogeneity and Other Pharmacological Properties over Conventional Heterogeneous ADCs. Mol. Pharm. 2015, 12, 3986–3998.
- Dennler, P.; Chiotellis, A.; Fischer, E.; Bregeon, D.; Belmant, C.; Gauthier, L.; Lhospice, F.; Romagne, F.; Schibli, R. Transglutaminase-based chemo-enzymatic conjugation approach yields homogeneous antibody-drug conjugates. Bioconj. Chem. 2014, 25, 569–578.
- Frutos, S.; Hernández, J.L.; Otero, A.; Calvis, C.; Adan, J.; Mitjans, F.; Vila-Perelló, M. Site-Specific Antibody Drug Conjugates Using Streamlined Expressed Protein Ligation. Bioconj. Chem. 2018, 29, 3503–3508.
- Yamazoe, S.; Kotapati, S.; Hogan, J.M.; West, S.M.; Deng, X.A.; Diong, S.J.; Arbanas, J.; Nguyen, T.A.; Jashnani, A.; Gupta, D.; et al. Impact of Drug Conjugation on Thermal and Metabolic Stabilities of Aglycosylated and N-Glycosylated Antibodies. Bioconj. Chem. 2022, 33, 576–585.
- Zhou, Q.; Kyazike, J.; Boudanova, E.; Drzyzga, M.; Honey, D.; Cost, R.; Hou, L.; Duffieux, F.; Brun, M.P.; Park, A.; et al. Site-Specific Antibody Conjugation to Engineered Double Cysteine Residues. Pharmaceuticals 2021, 14, 672.
- Benjamin, S.R.; Jackson, C.P.; Fang, S.; Carlson, D.P.; Guo, Z.; Tumey, L.N. Thiolation of Q295: Site-Specific Conjugation of Hydrophobic Payloads without the Need for Genetic Engineering. Mol. Pharm. 2019, 16, 2795–2807.
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106.
- Tian, F.; Lu, Y.; Manibusan, A.; Sellers, A.; Tran, H.; Sun, Y.; Phuong, T.; Barnett, R.; Hehli, B.; Song, F.; et al. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. USA 2014, 111, 1766–1771.
- VanBrunt, M.P.; Shanebeck, K.; Caldwell, Z.; Johnson, J.; Thompson, P.; Martin, T.; Dong, H.; Li, G.; D’Hooge, F.; Masterson, L.; et al. Genetically Encoded Azide Containing Amino Acid in Mammalian Cells Enables Site-Specific Antibody-Drug Conjugates Using Click Cycloaddition Chemistry. Bioconj. Chem. 2015, 26, 2249–2260.
- Zimmerman, E.S.; Heibeck, T.H.; Gill, A.; Li, X.; Murray, C.J.; Madlansacay, M.R.; Tian, C.; Uter, N.T.; Yin, G.; Rivers, P.J.; et al. Production of site-specific antibody-drug conjugates using optimized non-natural amino acids in a cell-free expression system. Bioconj. Chem. 2014, 25, 351–361.
- Li, X.; Nelson, C.G.; Nair, R.R.; Hazlehurst, L.; Moroni, T.; Martinez-Acedo, P.; Nanna, A.R.; Hymel, D.; Burke, T.R., Jr.; Rader, C.; et al. Stable and Potent Selenomab-Drug Conjugates. Cell Chem. Biol. 2017, 24, 433–442.e6.
- Okeley, N.M.; Toki, B.E.; Zhang, X.; Jeffrey, S.C.; Burke, P.J.; Alley, S.C.; Senter, P.D. Metabolic Engineering of Monoclonal Antibody Carbohydrates for Antibody-Drug Conjugation. Bioconj. Chem. 2013, 24, 1650–1655.
- Zhou, Q.; Stefano, J.E.; Manning, C.; Kyazike, J.; Chen, B.; Gianolio, D.A.; Park, A.; Busch, M.; Bird, J.; Zheng, X.; et al. Site-specific antibody-drug conjugation through glycoengineering. Bioconj. Chem. 2014, 25, 510–520.
- Li, X.; Fang, T.; Boons, G.J. Preparation of Well-Defined Antibody-Drug Conjugates through Glycan Remodeling and Strain-Promoted Azide-Alkyne Cycloadditions. Angew. Chem. Int. Ed. 2014, 53, 7179–7182.
- van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native mAbs Provides Homogeneous and Highly Efficacious Antibody-Drug Conjugates. Bioconj. Chem. 2015, 26, 2233–2242.
- Zhu, Z.; Ramakrishnan, B.; Li, J.; Wang, Y.; Feng, Y.; Prabakaran, P.; Colantonio, S.; Dyba, M.A.; Qasba, P.K.; Dimitrov, D.S. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. mAbs 2014, 6, 1190–1200.
- Tang, F.; Wang, L.X.; Huang, W. Chemoenzymatic synthesis of glycoengineered IgG antibodies and glycosite-specific antibody-drug conjugates. Nat. Protoc. 2017, 12, 1702–1721.
- Wijdeven, M.A.; van Geel, R.; Hoogenboom, J.H.; Verkade, J.M.M.; Janssen, B.M.G.; Hurkmans, I.; de Bever, L.; van Berkel, S.S.; van Delft, F.L. Enzymatic glycan remodeling-metal free click (GlycoConnect™) provides homogenous antibody-drug conjugates with improved stability and therapeutic index without sequence engineering. mAbs 2022, 14, 2078466.
- Strop, P.; Liu, S.H.; Dorywalska, M.; Delaria, K.; Dushin, R.G.; Tran, T.T.; Ho, W.H.; Farias, S.; Casas, M.G.; Abdiche, Y.; et al. Location matters: Site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013, 20, 161–167.
- Beerli, R.R.; Hell, T.; Merkel, A.S.; Grawunder, U. Sortase Enzyme-Mediated Generation of Site-Specifically Conjugated Antibody Drug Conjugates with High In Vitro and In Vivo Potency. PLoS ONE 2015, 10, e0131177.
- Wu, P.; Shui, W.; Carlson, B.L.; Hu, N.; Rabuka, D.; Lee, J.; Bertozzi, C.R. Site-specific chemical modification of recombinant proteins produced in mammalian cells by using the genetically encoded aldehyde tag. Proc. Natl. Acad. Sci. USA 2009, 106, 3000–3005.
- Rabuka, D.; Rush, J.S.; deHart, G.W.; Wu, P.; Bertozzi, C.R. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052–1067.
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde tag coupled with HIPS chemistry enables the production of ADCs conjugated site-specifically to different antibody regions with distinct in vivo efficacy and PK outcomes. Bioconj. Chem. 2014, 25, 1331–1341.
- Stevens, A.J.; Brown, Z.Z.; Shah, N.H.; Sekar, G.; Cowburn, D.; Muir, T.W. Design of a Split Intein with Exceptional Protein Splicing Activity. J. Am. Chem. Soc. 2016, 138, 2162–2165.
- Park, J.; Lee, Y.; Ko, B.J.; Yoo, T.H. Peptide-Directed Photo-Cross-Linking for Site-Specific Conjugation of IgG. Bioconj. Chem. 2018, 29, 3240–3244.
- Cheng-Sánchez, I.; Moya-Utrera, F.; Porras-Alcalá, C.; López-Romero, J.M.; Sarabia, F. Antibody-Drug Conjugates Containing Payloads from Marine Origin. Mar. Drugs 2022, 20, 494.
- Jin, Y.; Edalatian Zakeri, S.; Bahal, R.; Wiemer, A.J. New Technologies Bloom Together for Bettering Cancer Drug Conjugates. Pharmacol. Rev. 2022, 74, 680–711.
- Junutula, J.R.; Bhakta, S.; Raab, H.; Ervin, K.E.; Eigenbrot, C.; Vandlen, R.; Scheller, R.H. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J. Immunol. Methods 2008, 332, 41–52.
- Stimmel, J.B.; Merrill, B.M.; Kuyper, L.F.; Moxham, C.P.; Hutchins, J.T.; Fling, M.E.; Kull, F.C., Jr. Site-specific conjugation on serine right-arrow cysteine variant monoclonal antibodies. J. Biol. Chem. 2000, 275, 30445–30450.
- Voynov, V.; Chennamsetty, N.; Kayser, V.; Wallny, H.J.; Helk, B.; Trout, B.L. Design and application of antibody cysteine variants. Bioconj. Chem. 2010, 21, 385–392.
- Tumey, L.N.; Li, F.; Rago, B.; Han, X.; Loganzo, F.; Musto, S.; Graziani, E.I.; Puthenvant, J.; Casavant, J.; Marquette, K.; et al. Site Selection: A Case Study in the Identification of Optimal Cysteine Engineered Antibody Drug Conjugates. AAPS J. 2017, 19, 1123–1135.
- Shinmi, D.; Taguchi, E.; Iwano, J.; Yamaguchi, T.; Masuda, K.; Enokizono, J.; Shiraishi, Y. One-Step Conjugation Method for Site-Specific Antibody-Drug Conjugates through Reactive Cysteine-Engineered Antibodies. Bioconj. Chem. 2016, 27, 1324–1331.
- Dragovich, P.S.; Adhikari, P.; Blake, R.A.; Blaquiere, N.; Chen, J.; Cheng, Y.X.; den Besten, W.; Han, J.; Hartman, S.J.; He, J.; et al. Antibody-mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ERα). Bioorganic Med. Chem. Lett. 2020, 30, 126907.
- Dragovich, P.S.; Pillow, T.H.; Blake, R.A.; Sadowsky, J.D.; Adaligil, E.; Adhikari, P.; Bhakta, S.; Blaquiere, N.; Chen, J.; Dela Cruz-Chuh, J.; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 1: Exploration of Antibody Linker, Payload Loading, and Payload Molecular Properties. J. Med. Chem. 2021, 64, 2534–2575.
- Dragovich, P.S.; Pillow, T.H.; Blake, R.A.; Sadowsky, J.D.; Adaligil, E.; Adhikari, P.; Chen, J.; Corr, N.; Dela Cruz-Chuh, J.; Del Rosario, G.; et al. Antibody-Mediated Delivery of Chimeric BRD4 Degraders. Part 2: Improvement of In Vitro Antiproliferation Activity and In Vivo Antitumor Efficacy. J. Med. Chem. 2021, 64, 2576–2607.
- Dragovich, P.S. Degrader-antibody conjugates. Chem. Soc. Rev. 2022, 51, 3886–3897.
- Zhang, X.; Ou, C.; Liu, H.; Prabhu, S.K.; Li, C.; Yang, Q.; Wang, L.X. General and Robust Chemoenzymatic Method for Glycan-Mediated Site-Specific Labeling and Conjugation of Antibodies: Facile Synthesis of Homogeneous Antibody-Drug Conjugates. ACS Chem. Biol. 2021, 16, 2502–2514.
- Zhang, X.; Ou, C.; Liu, H.; Wang, L.X. Synthesis and Evaluation of Three Azide-Modified Disaccharide Oxazolines as Enzyme Substrates for Single-Step Fc Glycan-Mediated Antibody-Drug Conjugation. Bioconj. Chem. 2022, 33, 1179–1191.
- Shi, W.; Li, W.; Zhang, J.; Li, T.; Song, Y.; Zeng, Y.; Dong, Q.; Lin, Z.; Gong, L.; Fan, S.; et al. One-step synthesis of site-specific antibody-drug conjugates by reprograming IgG glycoengineering with LacNAc-based substrates. Acta Pharm. Sin. B 2022, 12, 2417–2428.
- Jevševar, S.; Kusterle, M.; Kenig, M. PEGylation of antibody fragments for half-life extension. Methods Mol. Biol. 2012, 901, 233–246.
- Humphreys, D.P.; Heywood, S.P.; Henry, A.; Ait-Lhadj, L.; Antoniw, P.; Palframan, R.; Greenslade, K.J.; Carrington, B.; Reeks, D.G.; Bowering, L.C.; et al. Alternative antibody Fab’ fragment PEGylation strategies: Combination of strong reducing agents, disruption of the interchain disulphide bond and disulphide engineering. Protein Eng. Des. Sel. PEDS 2007, 20, 227–234.
- Pepinsky, R.B.; Walus, L.; Shao, Z.; Ji, B.; Gu, S.; Sun, Y.; Wen, D.; Lee, X.; Wang, Q.; Garber, E.; et al. Production of a PEGylated Fab’ of the anti-LINGO-1 Li33 antibody and assessment of its biochemical and functional properties in vitro and in a rat model of remyelination. Bioconj. Chem. 2011, 22, 200–210.
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328.
- Zhou, C.; Lehar, S.; Gutierrez, J.; Rosenberger, C.M.; Ljumanovic, N.; Dinoso, J.; Koppada, N.; Hong, K.; Baruch, A.; Carrasco-Triguero, M.; et al. Pharmacokinetics and pharmacodynamics of DSTA4637A: A novel THIOMAB antibody antibiotic conjugate against Staphylococcus aureus in mice. mAbs 2016, 8, 1612–1619.
- Yu, S.; Pearson, A.D.; Lim, R.K.; Rodgers, D.T.; Li, S.; Parker, H.B.; Weglarz, M.; Hampton, E.N.; Bollong, M.J.; Shen, J.; et al. Targeted Delivery of an Anti-inflammatory PDE4 Inhibitor to Immune Cells via an Antibody-drug Conjugate. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 2078–2089.
- Lim, R.K.; Yu, S.; Cheng, B.; Li, S.; Kim, N.J.; Cao, Y.; Chi, V.; Kim, J.Y.; Chatterjee, A.K.; Schultz, P.G.; et al. Targeted Delivery of LXR Agonist Using a Site-Specific Antibody-Drug Conjugate. Bioconj. Chem. 2015, 26, 2216–2222.
- Pishesha, N.; Harmand, T.; Carpenet, C.; Liu, X.; Bhan, A.; Islam, A.; van den Doel, R.; Pinney, W., 3rd; Ploegh, H.L. Targeted delivery of an anti-inflammatory corticosteroid to Ly6C/G-positive cells abates severity of influenza A symptoms. Proc. Natl. Acad. Sci. USA 2022, 119, e2211065119.
- Maneiro, M.A.; Forte, N.; Shchepinova, M.M.; Kounde, C.S.; Chudasama, V.; Baker, J.R.; Tate, E.W. Antibody-PROTAC Conjugates Enable HER2-Dependent Targeted Protein Degradation of BRD4. ACS Chem. Biol. 2020, 15, 1306–1312.
- Pillow, T.H.; Adhikari, P.; Blake, R.A.; Chen, J.; Del Rosario, G.; Deshmukh, G.; Figueroa, I.; Gascoigne, K.E.; Kamath, A.V.; Kaufman, S.; et al. Antibody Conjugation of a Chimeric BET Degrader Enables in vivo Activity. ChemMedChem 2020, 15, 17–25.
- Ahn, G.; Banik, S.M.; Miller, C.L.; Riley, N.M.; Cochran, J.R.; Bertozzi, C.R. LYTACs that engage the asialoglycoprotein receptor for targeted protein degradation. Nat. Chem. Biol. 2021, 17, 937–946.
- Zhang, X.; Liu, H.; He, J.; Ou, C.; Donahue, T.C.; Muthana, M.M.; Su, L.; Wang, L.X. Site-Specific Chemoenzymatic Conjugation of High-Affinity M6P Glycan Ligands to Antibodies for Targeted Protein Degradation. ACS Chem. Biol. 2022, 17, 3013–3023.
- Kim, C.H.; Axup, J.Y.; Lawson, B.R.; Yun, H.; Tardif, V.; Choi, S.H.; Zhou, Q.; Dubrovska, A.; Biroc, S.L.; Marsden, R.; et al. Bispecific small molecule-antibody conjugate targeting prostate cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 17796–17801.
- Cui, H.; Thomas, J.D.; Burke, T.R., Jr.; Rader, C. Chemically programmed bispecific antibodies that recruit and activate T cells. J. Biol. Chem. 2012, 287, 28206–28214.
- Chapman, A.P.; Antoniw, P.; Spitali, M.; West, S.; Stephens, S.; King, D.J. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat. Biotechnol. 1999, 17, 780–783.
- Kawamura, A.; Miura, S.; Murayama, T.; Iwata, A.; Zhang, B.; Nishikawa, H.; Tsuchiya, Y.; Matsuo, K.; Tsuji, E.; Saku, K.; et al. Increased expression of monocyte CD11a and intracellular adhesion molecule-1 in patients with initial atherosclerotic coronary stenosis. Circ. J. Off. J. Jpn. Circ. Society. 2004, 68, 6–10.
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200.
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559.
- Negi, A.; Kesari, K.K.; Voisin-Chiret, A.S. Estrogen Receptor-α Targeting: PROTACs, SNIPERs, Peptide-PROTACs, Antibody Conjugated PROTACs and SNIPERs. Pharmaceutics 2022, 14, 2523.
- Garber, K. The PROTAC gold rush. Nat. Biotechnol. 2022, 40, 12–16.
- Dragovich, P.S.; Blake, R.A.; Chen, C.; Chen, J.; Chuh, J.; den Besten, W.; Fan, F.; Fourie, A.; Hartman, S.J.; He, C.; et al. Conjugation of Indoles to Antibodies through a Novel Self-Immolating Linker. Chemistry 2018, 24, 4830–4834.
- Ahn, G.; Banik, S.M.; Bertozzi, C.R. Degradation from the outside in: Targeting extracellular and membrane proteins for degradation through the endolysosomal pathway. Cell Chem. Biol. 2021, 28, 1072–1080.
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297.
- Zhou, Y.; Teng, P.; Montgomery, N.T.; Li, X.; Tang, W. Development of Triantennary N-Acetylgalactosamine Conjugates as Degraders for Extracellular Proteins. ACS Cent. Sci. 2021, 7, 499–506.
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-Pictet-Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconj. Chem. 2013, 24, 846–851.
- Goswami, R.K.; Bajjuri, K.M.; Forsyth, J.S.; Das, S.; Hassenpflug, W.; Huang, Z.Z.; Lemer, R.A.; Felding-Habermann, B.; Sinha, S.C. Chemically programmed antibodies targeting multiple alpha(v) integrins and their effects on tumor-related functions in vitro. Bioconj. Chem. 2011, 22, 1535–1544.
- Liu, Y.; Goswami, R.K.; Liu, C.; Sinha, S.C. Chemically Programmed Bispecific Antibody Targeting Legumain Protease and αvβ3 Integrin Mediates Strong Antitumor Effects. Mol. Pharm. 2015, 12, 2544–2550.
- Rader, C.; Sinha, S.C.; Popkov, M.; Lerner, R.A.; Barbas, C.F., 3rd. Chemically programmed monoclonal antibodies for cancer therapy: Adaptor immunotherapy based on a covalent antibody catalyst. Proc. Natl. Acad. Sci. USA 2003, 100, 5396–5400.
- Walseng, E.; Nelson, C.G.; Qi, J.; Nanna, A.R.; Roush, W.R.; Goswami, R.K.; Sinha, S.C.; Burke, T.R., Jr.; Rader, C. Chemically Programmed Bispecific Antibodies in Diabody Format. J. Biol. Chem. 2016, 291, 19661–19673.
- Lacek, K.; Urbanowicz, R.A.; Troise, F.; De Lorenzo, C.; Severino, V.; Di Maro, A.; Tarr, A.W.; Ferrana, F.; Ploss, A.; Temperton, M.; et al. Dramatic potentiation of the antiviral activity of HIV antibodies by cholesterol conjugation. J. Biol. Chem. 2014, 289, 35015–35028.
More