1. Molecular Formats: Structures and Properties
Monoclonal antibodies are the most often used proteins to construct immunocytokines since they demonstrate high affinity, selectivity, and well-predicted and studied properties and characteristics. They consist of two heavy (Hc) and two light (Lc) chains forming two Fab arms (V
LC
LV
HC
H1) and a Fc-region ([C
H2C
H3]
2). The main function of the Fc-region is to interact with proteins of the complement system and Fc receptors, including neonatal Fc receptor (FcRn), which plays an important role in antibody recirculation and maintaining a long serum half-life
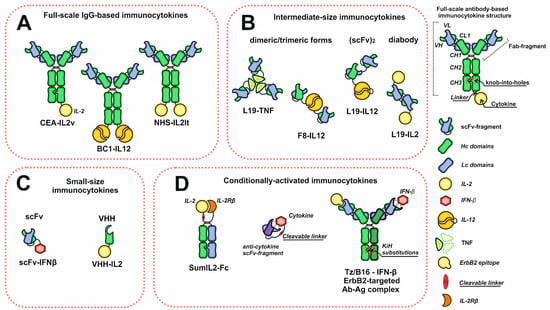
[1][2][26,27]. A wide variety of existing antibodies with a broad range of actual and possible therapeutic targets made antibodies extremely convenient objects for devising immunocytokines. Most antibody–cytokine fusion proteins can be classified into three groups: full-scale IgG fusion proteins and intermediate- and small-size molecules based on antibody fragments with binding activity (
Figure 1). One of the most common format choices, in case one wishes to achieve the long serum half-life, is a recombinant full-length IgG antibody with a C-terminus-linked cytokine. This recombinant molecule retains all IgG inherent activities, but is bivalent against cytokine receptors, which could be adverse if cytokine–receptor interactions are too strong. This may lead to immunocytokine trapping by the excess of receptors and the resulting lack of necessary concentrations at the target site
[3][28]. One way to overcome this problem is to introduce knob-into-holes
[4][29] or other heavy chain heterodimerization platform mutations into heavy chain genes in order to retain the bivalency and avidity of the antibody, but to decrease the number of cytokine receptor binding moieties to the number of one. Another solution is to switch to smaller formats, such as Fab (antigen-binding fragment) or scFv fragments (single-chain variable fragment). However, antibody fragments are characterized by different pharmacokinetic properties with shorter serum half-life and increased diffusion capacity if compared to the full-scale IgG
[5][6][30,31]. On the contrary, rapid clearance and diminished tumor retention are the drawbacks of intermediate- and small-sized formats, which cause low absolute accumulation about one fold lower than full-length IgG. By contrast, the decrease in serum half-life may be beneficial in terms of keeping blood concentrations low to reduce side effects.
Figure 1. Schematic representation of the diversity of immunocytokine formats. (A) Immunocytokines based on full-scale IgG. (B) Intermediate-size immunocytokines. (C) Small-size immunocytokines based on antibody scFv or VHH fragments. (D) Conditionally activated immunocytokines. Abbreviations: VH and VL—heavy and light chain variable domains; CL—light chain constant domain; CH1, CH2, CH3—heavy chain constant domains; scFv—single chain variable fragment; VHH—single domain antibodies.
2. Mechanisms and Modes of Action
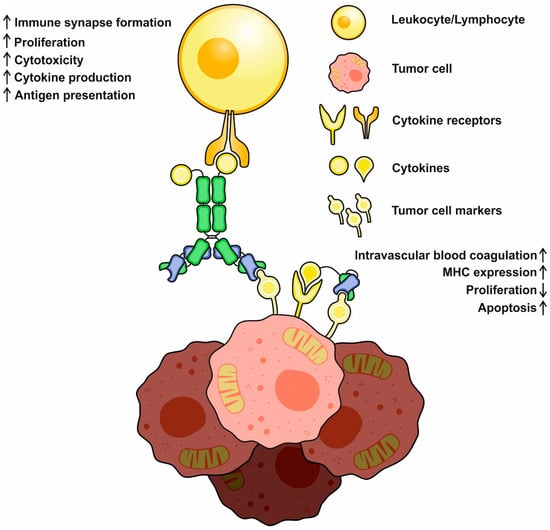
Since immunocytokines consist of the antibody and cytokine moieties, their mechanisms of action include the inherent ones typical of both antibodies and cytokines, with some new properties emerging. Full-scale IgG-based immunocytokines retain CDC (complement-mediated cytotoxicity) and ADCC (antibody-dependent cellular cytotoxicity) activities without any significant decrease compared to the prototype antibodies
[7][8][45,46]. Thus, they directly bind to tumor cells and trigger those reactions. However, the concentrations of immunocytokines used during therapies are one or two orders of magnitude lower than those of anti-cancer monoclonal antibodies, which makes ADCC and CDC as a mechanism controversial and rather additional than principal. Moreover, some immunocytokines undergo site-specific mutagenesis with a purpose to disrupt Fc and C1q binding, and thus improve localization and decrease off-target effects
[9][47]. Being a delivered molecule, a cytokine plays a more important role in antibody–cytokine fusions. Distinct cytokines may activate different subsets of leukocytes and lymphocytes and induce their proliferation or infiltration at the site of the tumor. Pro-inflammatory payloads increase NK- and T-lymphocyte killing capability, but some of them (e.g., IL-2) enhance immunosuppressive properties of T
regs (regulatory T cells), mostly an unwanted effect during cancer treatment
[10][48]. Targeted cytokine-mediated activation of the immune cells may trigger massive cytokine production at the disease site, making the local environmental conditions more favorable for local immune system cell functioning.
The delivered cytokines can also directly affect tumor cells and decrease their proliferation and motility or increase MHC I expression and antigen presentation, making a tumor more recognizable for immune cells and less capable of metastatic spread. Some of the cargoes (e.g., TNF-α or TRAIL) can directly induce apoptosis through ligand–receptor interactions, and therefore, could be considered as “magic bullets”. Tumor vasculature is also an important target for cytokines; for example, TNF-α, IL-1, and IL-6 were reported to promote intravascular blood coagulation of small capillaries
[11][49]. Finally, immunocytokines can act similarly to bispecific antibodies, establishing the crosslinks between tumor and immune system cells (
Figure 2). Interactions mediated by IL-2-based antibody–cytokine fusion proteins induce polarization, increase cell adhesion, and activate immune synapse formation between tumor and IL-2R+ NK cells, facilitating their cytotoxic functions
[12][50]. Immunocytokines were reported to redirect FcR-lacking cells, e.g., effector T cells, to malignancies through the formation of stable conjugates between cell membranes. After establishing these contacts, T cells mediate Fas-Fas-dependent lysis, followed by consequent tumor cell death
[13][51].
Figure 2. A schematic representation of basic mechanisms of immunocytokine action. Cytokines may activate and redirect immune system cells to tumor cells and initiate immune synapse formation. Activated immune cells increase (↑) in cytotoxicity, cytokine production, and proliferation. Direct action of cytokines decreases (↓) tumor cell propagation, increases MHC expression and antigen presentation, and might activate proapoptotic pathways.
3. IL-2-Based Immunocytokines
Interleukin-2 (IL-2) is a 15–16 kDa proinflammatory cytokine known and discovered as a T-cell growth factor
[14][52]. IL-2 has multiple functions: it promotes the growth and development of peripheral immune cells, and it is essential for Th
9 generation and T
reg-cell differentiation, but it also demonstrates an inhibitory effect on Th
17 and T
FH differentiation. There are three different classes of IL-2 receptors with different affinities, consisting of the IL-2Rα (K
D~10
−8 M) subunit only; the IL-2Rβ and IL-2Rγ subunits (K
D~10
−9 M); and the IL-2Rα, IL-2Rβ, and IL-2Rγ subunits (K
D~10
−11 M). Whereas intermediate-affinity receptors are expressed mostly on resting NK and T cells, high-affinity receptors are typical of lymphocytes after their activation, making them more sensitive to this cytokine. IL-2 is also able to improve the tumor-killing capacity of different immune system cells by increasing the cytolytic activity of NK cells and lymphokine-activated killer cells and inducing the rapid proliferation of CD8+ T cells
[15][53]. Using IL-2 in clinical practice (Aldesleukin) started three decades ago and was considered as the first effective immunotherapy for human cancer
[16][4].
Further studies demonstrated that IL-2 therapy may be successfully combined with adoptive cellular therapy or genetically activated T-cells administration, resulting in a statistically significant increase of survival rate, as well as other clinical indicators
[17][54]. However, the limited success of IL-2 therapy might be explained by a narrow therapeutic index, vascular leakage syndrome, and toxicities, which make IL-2 more useful combined with chemotherapy
[18][55]. Other facts presented below demonstrate that IL-2 has a few protumorigenic properties, which makes its use for cancer treatment less encouraging. For example, IL-2 is an inducer of Th
9-cell differentiation and Th
9-mediated IL-9 production, two factors correlating with better tumor cell survival and apoptosis resistance
[19][20][56,57]. IL-2 inhibits the differentiation of Th
17 cells, a population of lymphocytes performing an important function in protecting the organism from exogenous pathogenic fungi and bacteria that makes patients less resistant to infections during therapy. IL-2 was also shown to be crucial for the suppressive activity of peripheral and thymus-derived T
reg cells, normally responsible for self-tolerance during tumorigenesis stimulation, and infiltration of peripheral T
reg into tumors may inhibit antitumor immunity
[21][58]. Finally, IL-2 is characterized by a serum half-life limited to ≈7 min
[22][59]. All the facts
we have discussed thus far, as well as other minor factors, led scientists to develop new methods of overcoming IL-2 toxicity, side effects, and limitations.
The restrictions related to limited serum half-life and T
reg activation could be overcome by IL-2 engineering, making fusion proteins, or chemical conjugation with PEG. Nonetheless, none of these techniques dramatically increases the specificity of the cytokine to tumor cells
[23][60]. One of a few methods used to overcome the negative immunosuppressive effects of IL-2 is IL-2 receptor–cytokine binding interface engineering performed to decrease the binding to high-affinity T
reg interleukin receptors. IL-2 with abrogated IL-2Rα (CD25) binding or enhanced IL-2Rβ binding was demonstrated to induce less efficient T
reg expansion and tumor infiltration, with more efficient tumor-specific CD8+ T-cell proliferation
[9][24][47,61]. A no-α-designed mutant form of IL-2 with reduced binding capacity to IL-2Rα showed a greater therapeutic efficacy and anti-metastatic effect in a mouse metastasis model than wild-type IL-2. This could be explained by the preferential stimulation of NK and CD8+ T cells, along with the avoidance of stimulation of CD4+F FoxP3 T
regs [25][62]. However, both the toxicity and lack of tumor specificity represent a serious limitation for harnessing the engineered IL-2 forms. Therefore, IL-2 and tumor-specific antibody fusions are widely constructed and tested.
Quite a different tumor marker was selected during Selectikine (NHS-IL-2
Lt, EMD521873) development, which consists of an IL-2 low-toxicity mutant (D20T) fused at the N terminus to a humanized NHS antibody targeted against DNA and histone complexes released at the necrotic core of tumors
[26][81]. In syngeneic mouse metastatic model experiments, intravenous administration of 80 mcg/injection of NHS-IL-2
Lt for 5 days
[27][63] dramatically reduced the metastatic load in lungs and liver. NHS-IL-2
Lt has already completed Phase I of clinical studies and demonstrated preliminary data of inefficacy
[28][24]. Patients with different types of cancer tumors (colorectal, ovarian, prostate, and renal cancers) were divided in groups, and Selectikine at 0.15, 0.3, and 0.45 mg/kg was administered intravenously every 3 weeks during 3 consecutive days. The treatment was followed by a strong activation of T cells and a minor activation of NK cells. Unfortunately, only disease stabilization was observed in a few patients. However, these results may have been obtained due to the heterogeneity of the tumors in the patient groups and the considerable influence of prior treatment
[29][30][74,82].
4. IL-12-Based Immunocytokines
IL-12 is a 70 kDa pro-inflammatory cytokine modulating NK- and T-cell activities. Its biologically active form is produced by monocytes, neutrophils, dendritic cells, and B cells in response to pathogen stimulation. This form comprises p35 and p40 proteins linked by a disulfide bridge. Anti-tumor and antimetastatic activities of IL-12 were exploited in several preclinical models
[31][83], but during clinical studies, IL-12 administration failed to elicit any sustained anti-tumor response because of several toxicities, adverse effects, and the increase in tumor immunosuppressive properties by IL-10 induction
[32][84]. In light of the above, the untargeted administration of IL-12 is not pursued nowadays or in the future, but targeted delivery of IL-12 combined with other therapeutics seems to be rather promising. Experiments performed on mouse models demonstrated that tumor-specific delivery of IL-12 increases the number of T lymphocytes, macrophages, and NK cells infiltrating the tumor and decreases the angiogenesis
[33][85]. According to Pasche and coauthors, the format of immunocytokine greatly impacts the effectiveness of the fusion protein if the cytokine molecular weight is high or it consists of two or more subunits, e.g., IL-12
[34][86]. After failing to localize an F8-based IL-12 diabody-based immunocytokine (average M
w 190 kDa) at the tumor site, they decided to use a ScFv platform for fusing the construction. Two recombinant proteins made up of identical scFv fragments and p35 or p40 subunits (
Figure 1) were used for IL12-F8 heterodimer formation (scFv
F8-p35/p40-scFv
F8) in order to localize IL-12 at fibronectin extra domain A-expressing tumors. In a mouse model with animals bearing F9 large subcutaneous tumors (150–250 mm
3) among the tested molecules only, IL12-F8 was able to mediate significant growth retardation after a single-dose injection (6 mkg). Immunohistochemistry analysis of the tumors revealed a massive tumor infiltration by NK cells and leukocytes and a mild increase in CD4+ T cells. During experiments, animal serum levels of IFN-γ were altered, which is important for tumor angiogenesis inhibition by CD4+ T cells
[35][87]. Better anti-tumor effects were observed during co-administration of IL12-F8 with chemotherapy. A combination of IL12-F8 with paclitaxel gave encouraging results, with complete tumor eradication in all animals in A20 lymphoma models, and achieving 50% tumor-free animals in the tested sample in F9 teratocarcinoma models
[34][86]. Another immunocytokine, L19-IL12, which is specific to fibronectin EBD, demonstrated an anti-tumor effect very similar to IL12-F8. L19-IL12 is an L19 scFv-based fusion protein containing both p40 and p35 interleukin subunits separated by the flexible linker sequence. In C51 colon-carcinoma-bearing mice, 2.5 mcg injections of L19-IL12 every 48 h for 13 days suppressed tumor growth in the experimental group to the weight of 0.31 g, compared to 1.45 g in the control group. Even superior inhibitory effects were obtained in a F9 murine teratocarcinoma model in 129Sv mice in animals treated with L19-IL12, with the final tumor weight equal to 0.11 g versus 1.45 g in the non-treated group. Further experiments proved the favorable tumor-targeting properties of L19-IL12 recombinant proteins with a tumor–to–blood ratio over 31 and an apparent serum half-life of nearly 22 h
[36][88].
Nowadays, only two IL-12-based immunocytokines have been progressed to clinical trials: NHS-IL12 and BC1-IL12. NHS-IL12 is a full-scale IgG with a C-terminus-fused IL-12 p35 subunit via the glycine–serine linker. The other subunit p40 is translated separately and forms a heterodimer with p35 by a disulfide bond. Compared to systemic administration of IL-12, NHS-IL12 demonstrated reduced toxicities due to a targeted delivery of the molecule to the necrotic core of the tumor. A murine version of NHS-IL12 (NHS-murIL12) exerts moderate dose-dependent anti-tumor effects on subcutaneous tumor growth, even after a single injection in animal models. Animals treated with NHS-muIL12 show increased MHC I expression on dendritic cells and proliferation of CD49B+ NK, and CD8+ T cells display stronger p15E-specific CD8+ T-cell response and altered IFN-γ serum levels. Despite the difference in the molecular weights of IL12 and NHS-muIL12, the latter demonstrated a tumor-retention effect superior to recombinant muIL-12
[37][89], which enhanced and prolonged cytotoxic effector functions of CD8+ T cells at the tumor site. The cured mice did not develop any tumors after a repeated administration of the same tumor cell line, which indicates the vaccination effect of the treatment. A fundamental advantage of NHS-muIL12 is an increase in local tumor IL-12 concentration that seems to be important to bypass the immunosuppressive tumor microenvironment and fully activate tumor-specific T cells
[28][24]. A combination of NHS-IL12 and anti PD-L1 antibody avelumab demonstrated a synergistic anti-tumor effect in MC38 and MB49 tumor-bearing C57BL/6 mice and seems to be promising for tumor cotreatment
[38][90]. Currently, NHS-IL12 has already completed phase I clinical trials, and it was consistently well tolerated, with preclinical data suggesting its potential to improve anti-tumor responses with other standard treatments
[39][40][91,92]. Another immunocytokine, BC1-IL12, finished phase I clinical trials for renal cell carcinoma and malignant melanoma treatment and proceeded to clinical trials phase II. BC1-IL12 is a recombinant protein consisting of humanized BC1 mAb targeting EDB-containing fibronectin (different epitope from L19)
[40][92] and a C-terminus-fused p35 IL-12 subunit with a representative structure similar to NHS-IL12. During experiments in xenogeneic mice with subcutaneous tumors, BC1-muIL12 succeeded in decreasing the number of metastases. In a PC3 lung cancer model in SCID mice lacking any functional T and B cells, 16 mcg injections of BC1-muIL12 daily were enough to completely prevent further tumor outgrowth
[41][93]. The maximum tolerated dose (MTD) in cynomolgus monkeys determined for 8 weeks was 2.5 mcg/kg, which is 10-fold higher than the corresponding value for huIL-12. In human patients, the MTD was defined as 15 mcg/kg, and the serum half-life was approximately 22 h. During clinical studies, some grade 2 toxicities were registered, but, overall, were lower than those reported for IL-12 as a monotherapy agent
[42][43][94,95].
5. TNF-Based Immunocytokines
Tumor necrosis factor alpha (TNF-α) was discovered back in 1975 as a factor released from host cells, causing hemorrhagic necrosis of sarcoma Meth A and other tumors
[44][96]. TNF-α is primarily synthesized by immune cells as a 34 kDa transmembrane protein with a 17 kDa extracellular domain. After being cleaved by TACE/ADAMS17 proteinase, its soluble form is released and forms homotrimers capable of interacting with TNF receptors
[45][97]. The roles of TNF-α in carcinogenesis are quite controversial. On the one hand, TNF-α promotes inflammation, regulates cell survival and immunosuppression, and can induce invasion of neoplastic cells into the surrounding tissues
[46][98]. All these processes corelate with poor prognosis and ultimately complicate cancer treatment. On the other hand, TNF-α responsible for programmed cell death activation exerted promising anti-tumor effects in several experiments
[47][99]. Considering the above, it is most logical that TNF-α is being investigated as a potent anti-tumor agent. However, high toxicity is a serious limiting factor, which imposes restrictions on the systemic use of this cytokine, with limb perfusion as a single effective method
[48][100].
Taking into account the satisfactory tumor–to–blood ratio of many immunocytokines, the appearance and development of TNF-based antibody fusion proteins seems only natural. Because of the trimeric structure of active TNF-α, using full-length IgG will lead to the formation of immunocytokine complexes with extremely high molecular weights above 500 kDa, with a very limited diffusion capacity. That is why antibody fragments (Fab or scFv) and minibodies are the most convenient vehicles for TNF-α delivery. A fully human L19 antibody is one of the most often used molecules to construct TNF-based antibody–cytokine fusion proteins. In BALB/C mice with implanted WEHI-164 cells L19-mTNF, an immunocytokine consisting of L19 antibody fragment and murine TNF-α demonstrated mild tumor growth inhibition as a single agent, with complete response in one of four animals. The combination of L19-mTNF and trabectedin enhanced this effect to three of four complete responses. Another chemotherapeutic, decarbazine, as a single agent was capable of eradicating tumors in three out of four mice, but while administered with L19-mTNF, the number of complete responses increased to four of four. Very similar synergistic effects were observed for L19-mTNF and anti-PD-1 antibody
[49][101]. Anti-tumor effects mediated by antibody-based TNF-α delivery could also be increased by a proper combination with other cytokines, e.g., IL-2. Menssen and coauthors reported that in their multiple myeloma model experiments with BALB/C J558L mice, a combination of two immunocytokines, L19-TNFα and L19-IL2, yielded the most effective results, as 75% of animals responded to the therapy, and 58% achieved complete tumor eradication. Treatment with either L19-TNFα or L19-IL2 was less efficacious, leading to complete tumor eradication in 42% or 25% mice, respectively
[50][102]. TNF-based immunocytokines also display a very pronounced cytotoxic effect against the endothelial cells, which may be a cause of tumor vessel lesions. This speculation is confirmed at least by the properties of TA99-TNFα, an immunocytokine that consists of a TA99 scFv fragment (anti gp75 melanoma antigen) and TNF-α moieties. TA99-TNFα was able to kill fibroblasts and endothelial cells, but displayed minimal activity against B16 melanoma cells, in contrast to TA99 IgG2a antibody. In vivo TA99-TNFα injections (7 mcg) boosted the influx of NK cells and macrophages into B16 lesions and caused tumor necrosis. According to Murer
[51][103], a combination of TA99-TNFα and TA99 antibody could be an effective remedy, either for tumor treatment or metastasis prevention. Another immunocytokine, F8-TNFα, an analogue of L19-TNFα, was also reported to induce intravascular coagulation of tumor blood vessels. In a sarcoma mouse model, a combination of doxorubicin (5 mg/kg) and F8-TNFα (2 mg) was able to completely eradicate an established 70 mm
3 tumor, a result unachievable by a single-agent treatment
[52][104].
6. Interferon-Based Immunocytokines
Initially, interferons were discovered and characterized for their antiviral properties. Meanwhile, in the context of cancer, they exhibit several direct and indirect therapeutic effects
[53][107]. Among all interferons (IFN), only IFN-α, IFN-β, and IFN-γ are exploited for immunocytokine construction. Interferon alpha (IFN-α) is approved by the FDA for treating patients with high-risk melanoma, follicular lymphoma, hairy cell leukemia, and chronic myelogenous leukemia and as the first-line treatment drug for renal cell carcinoma. Using IFN-α in humans polarizes immune responses towards Th1, increases NK cytotoxicity and survival, and enhances the propagation of cytotoxic T lymphocytes and the maturation of dendritic cells. For some cancer types, an anti-angiogenic effect on tumor vasculature and the ability of IFN-α to induce caspase-dependent apoptosis were observed
[54][108]. Unfortunately, systemic administration of IFN-α is limited by a short therapeutic window and the lack of any specificity to tumors and malignancies. In contrast, IFN-α-based immunocytokines demonstrate good targeting properties. For example, fusion of IFN-α to anti-CD38 mAb increased cytokine specificity to CD38+ tumor cells 10,000-fold, meaning that patients might be safely treated with high doses of fusion protein
[55][109]. After injection, IFN-α–based immunocytokines tend to localize at the tumor microenvironment and demonstrate good tumor–to–blood ratios close to 21:1
[56][110]. Reducing the severity of off-target effects might be achieved by an additional approach aimed at IFN-α engineering. Replacing wild-type IFN-α with an engineered, attenuated version (att) with the capability of inducing proinflammatory markers decreased the toxicity of CD38-(att)IFN-α for animals 100-fold, without substantial loss of anti-tumor activity. Despite using an attenuated version of interferon, anti-CD38-(att)IFN-α sustained significant anti-tumor activity in various cell lines and was capable of completely eradicating the established NCI-H929 subcutaneous tumors in all tested mice
[55][109]. Although IFN-α-based immunocytokines display pronounced anti-tumor and anti-proliferative effects, some data suggest that they might be implemented on tumor vasculature through a direct action. Despite tumor-specific accumulation of scFv(F8)-IFNα in two EDA+ tumor models and a pronounced antitumor effect, immunohistochemical staining revealed the preferential localization of the immunocytokine close to endothelial and perivascular cells at different injection administration doses. In addition, from a safety perspective, scFv(F8)-IFNα was well tolerated in mice, even in repeated 150 mcg injections
[56][110]. In syngeneic mouse tumor models resistant to anti-PD-L1 therapy, IFN-α full-scale IgG-fusion protein infusions, in contrast to IFN-α alone, were tolerated well and were capable of three-fold tumor volume reduction compared to the control group
[57][111]. Notably, these results demonstrate that IFN-α fusion proteins may well be considered for treating patients resistant to anti-PD-L1 therapy. The effectiveness of IFN-α-based immunocytokines strongly depends on in vivo models, one of the most sensitive being the Daudi xenograft model. Immunocytokine 20-2b, comprised of IFN-α2b moieties and CD20-targeting IgGs, shows enhanced ADCC compared to veltuzumab and increases the median survival time in SCID mice in an early Daudi xenograft model by more than 100 days over saline and veltazumab groups given even a single dose
[58][112]. The recombinant molecule (C2-2b-2b), with a similar format targeted at human leukocyte antigen (HLA-DR), demonstrates enhanced apoptosis-prompting ability in multiple myeloma and lymphoma cells, compared to 20-2b and the mixture of anti-HLA-DR mAb and IFN-α2b. In the Daudi model, C2-2b-2b significantly increased the survival time after 1 mcg injections, compared with other tested agents. For both anti-CD20 and HLA-DR immunocytokines, the mice survival correlated with their in vitro cytotoxicity, standing in the subpicomolar IC
50 range. Although in vivo the benefit of C2-2b-2b was superior to its cognate anti-CD20 fusion protein, C2-2b-2b specificity also caused elevated toxicity against healthy peripheral blood mononuclear cells
[59][113], a fact mentioned as a side effect. A bispecific variant constructed based on these two immunocytokines exhibited an even greater anti-tumor effect in Daudi Lymphoma than its monospecific prototypes
[8][46]. However, the full potential of the conjugates has not yet been demonstrated due to the low sensitivity of murine cells to human interferon.
Interferon beta (IFN-β) is not extensively used for anti-cancer therapeutic development, though some properties of this cytokine imply that it could be used for potent treatment. One of many IFN-β antitumor mechanisms consists of cell S-phase cycle arrest
[60][114]. IFN-β also downregulates c-myc expression, one of few protooncogenes
[61][115]. An increase in Fas, FasL, and TRAIL expression induced by IFN-β may be exploited for targeted cas8-dependent apoptosis mediation
[62][116]. In addition, angiogenesis in tumors is highly suppressed by IFN-β, even at low doses
[63][117]. In vitro studies confirmed that the direct killing effect of IFN-β-based immunocytokines is likely to depend more on interferon activity than on antibody activity
[64][118]. In vivo ErbB2-targeted IFN-β demonstrates considerable tumor growth suppression in humanized mouse gastric cancer xenograft models in a direct manner. Tumor growth inhibition was accompanied by a local 2.5-fold increase in CD8+ T cells and a 2.4-fold decrease in T
regs compared to the vehicle control
[30][82]. Interestingly, in vivo IFN-β fusion protein suppresses tumor growth mostly by inducing host immune responses
[65][119]. The half-life of IFN-β within full-scale-based immunocytokines increases from 88 min to 15 h, making it more convenient for administration during treatment. Although in mice
[64][65][118,119], human IFN-β-based immunocytokines are well tolerated, this conclusion could not be directly extrapolated to humans due to the low sensitivity of the animals to human interferon. Therefore, its toxicity would still be a serious challenge to overcome.
Delivering interferon to a lesion in a safe manner might be achieved in a neutralized state with a few strategies. One of the strategies is to deliver interferon by a bispecific antibody targeted to the tumor cell marker and capable of binding and neutralizing the interferon with a second arm, thus disrupting interferon–receptor interactions during transportation, which may cause potential side effects. Due to high molecular weight, the molecular structure comparable to IgG and the active Fc-part of this complex is stable and might sustain satisfactory serum persistence
[66][67][44,120]. Another strategy uses latency-associated protein of TGF-β1 fused to IFN-β via a matrix metalloproteinase labile linker, which is responsible for masking interferon from receptor interactions. At the tumor site, matrix metalloproteinases cleave the linker with a following release of interferon in its active form. The ‘latent’ cytokine has a 37.6 times longer half-life than was reported for IFN-β alone and, thus, could be administered systematically at lower doses
[68][43].
Interferon gamma (IFN-γ) is a type II interferon produced by NK cells and activated cytolytic T cells. IFN-γ participates in macrophage activation, increases their oxidative metabolism, and enhances tumor cell killing. Furthermore, IFN-γ is responsible for the augmentation of MHC expression, which enhances the tumor cell immunogenicity
[69][121]. For different types of cancer, IFN-γ-mediated antiproliferative, antiangiogenic, and proapoptotic effects were discovered, and some clinical studies were reported
[70][71][72][122,123,124]. IFN-γ targeted to fibronectin EDA, EDB, or CD70 demonstrated superior antitumor activity, compared to non-specific IFN-γ, in distinct models: sarcoma, teratocarcinoma, and lung cancer. The increase in the number of infiltrating macrophages and T and NK cells was registered by immunofluorescence analysis of histological tumor tissue samples in IFN-γ-treated animal groups. Targeted delivery of IFN-γ resulted in tumor microenvironment and metastasis control, compared to non-fused immunocytokine moieties. However, partial immunocytokine sequestration by other tissues containing IFN-γ receptors was observed during experiments. Additional IFN-γ administration resulted in competition for receptors with immunocytokines, and they partially resolved that problem without any apparent influence on animal body weight and negligible toxic effects
[73][74][75][125,126,127].
7. Other Immunocytokines
In addition to the cytokines listed above, various other ones have been investigated for the construction of antibody fusion proteins. IL-1β and IL-6 fused to anti-fibronectin EDA diabody were constructed and tested in a murine F9 teratocarcinoma model, exhibiting 10% and 5% animal weight loss at 5 and 225 mg dose, respectively. Unfortunately, only 50% tumor growth rate inhibition was observed, which is substantially lower than for other proinflammatory immunocytokines (e.g., F8-TNF-α)
[76][128]. Another cytokine with a potent antitumor effect was used for F8-mIL7-F8 construction, a molecule comprising two anti-fibronectin EDA scFv fragments and murine IL-7. This recombinant protein exhibited good tumor targeting, with tumor–to–blood ratio being 16:1 24 h post injection, and it dramatically inhibited F9 tumor growth in mice. The combination of F8-mIL7-F8 and paclitaxel improved this therapeutic effect, compared to the monotherapy treatment
[77][129]. Tumor-targeting properties of distinct fusion proteins in special cases may greatly depend on the glycosylation profiles of cytokines. A diabody format immunocytokine, F8-IL-9, produced in transient CHO cell culture was capable of localizing at F9 teratocarcinoma lesions, but after establishing the stable cell line, failed to do so. IL-9 has four O-glycosylation sites; thus, the difference in targeting two identical proteins was explained by the difference in glycosylation in stable and transient cell cultures
[78][130]. However, further research of F8-IL-9 showed the futility of F8-IL-9 anti-cancer treatment, but its potency for pulmonary hypertension treatment was discovered
[79][131].
IL-15 plays different roles in immune regulation, but also may act as a proinflammatory cytokine. In cancer models, IL-15 acts in the same manner as IL-2, being less toxic and considered as a promising cytokine for metastasis treatment. In an EL4 GD2+ lymphoma tumor model, anti-GD2-IL-15 significantly inhibited tumor development and increased mice mean survival, whereas administration of anti-GD2 diabody and IL-15 produced no significant effects. The efficacy of recombinant anti-GD2-IL-15 was proved in a liver metastasis NXS2 cell model, where this immunocytokine totally inhibited the development of metastasis. The bioavailability of anti-GD2-IL-15 was higher than that of IL-15, and its serum half-life increased 30-fold
[80][132]. Another IL-15-based immunocytokine, comprised of IL-15 and full-size anti-PD-L1 antibody (srKD033), showed a robust antitumor effect with even a single dose in several diverse syngeneic murine tumor models. In a CT26 murine colon cancer model, a single dose of srKD003 produced durable antitumor immunity and resistance to challenges. The retention of srKD003 in the tumor microenvironment facilitated IL-15-dependent cytotoxic cell expansion, activation of T and NK cells via PD-L1 inhibition, and IL-15 stimulation
[81][133]. Although srKD003 has limited application for tumors resistant to immune checkpoint inhibitors, its cognate, IL-15-based PD-L1-targeted immunocytokine LH01, can overcome primary resistance to PD-1/PD-L1 blockade by down-regulating TGF- levels within a tumor microenvironment, with no significant effect on PD-L1 expression levels. Additionally, it can induce local inflammation around malignancies, enhance CD8+ lymphocytes and NK-cell infiltration, and decrease the number of local T
reg populations
[82][134].
At least one publication focuses on IL-17-derived immunocytokines. In this study, IL-17 was fused to the C-terminus of a scFv fragment of fibronectin EDA-specific F8 antibody to investigate the anti-tumor effects of the construct. The construct (F8-IL-7) was capable of binding both IL-17 receptors and fibronectin EDA in vitro. In vivo, F8-IL-17 stimulated angiogenesis and leukocyte infiltration in mice with subcutaneous F9 tumors, but no significant inhibition of tumor growth was observed
[83][135]. Yet, more promising results were obtained with another proinflammatory cytokine, IL-21, a pleiotropic type I cytokine mainly produced by T and NK cells
[84][136]. Using IL-21-based immunocytokines to improve anti-PD-1/PD-L1 therapy, as the lack of efficient T-cell activation may be responsible for low response rates to checkpoint blockaders in different cancer treatments, seems promising. Thus, an anti-PD-1 antibody may be used as a vehicle for delivering IL-21 to reactive T cells and, therefore, enhance their cytolytic properties. A fusion protein, PD-1Ab21, which comprises anti-PD-1 diabody and two IL-21 molecules, proved this assumption and showed potent antitumor effects in mice with established tumors. This effect was accompanied by an expansion of tumor-specific CD8+ T cells, and it increased the frequency of memory stem T cells superior to infusions of IL-21 combined with PD-1 blockader
[85][137]. In humanized mice refractory to anti-PD-1, this approach provided significant protection
[86][138].