Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Abu Saim Mohammad Saikat and Version 1 by Abu Saim Mohammad Saikat.
- autophagy
- cancer
- autophagy inhibitors
- autophagy activators
- autophagic cell death
1. Targeted Drug for Autophagy Activation in Cancer Therapy
In cancer, autophagy helps promote and inhibit tumors by helping or hindering cancer cells from growing and dividing, while some drugs that fight cancer control autophagy; therefore, chemotherapy controlled by autophagy can cause cancer cells to live or die [30]. Autophagy regulation also affects the expression of proteins that promote or inhibit tumor growth [31], and the mechanism of action associated with autophagy stimulation is presented in Table 1. Additionally, the stimulation of autophagy by various chemicals and medications in cancer treatments is presented in Figure 3.
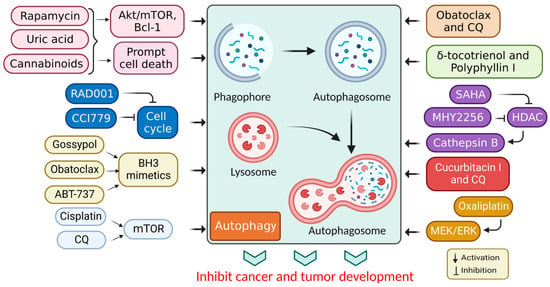
Figure 3. Autophagy stimulation by several compounds and drugs used in cancer therapy.
Table 1. Recently used drugs for autophagy stimulation and their mechanisms of action.
| Drug | Model | Molecular Mechanism | Reference |
|---|---|---|---|
| Azithromycin | Non-small-cell lung cancer (NSCLC) cells | Lysosomal membrane permeabilization for apoptosis induction | [32] |
| Penfluridol | Renal cell carcinoma | Induction of autophagy-mediated apoptosis and stemness inhibition | [33] |
| Canagliflozin | Acute kidney injury (AKI) | Cisplatin-mediated AKI by activating AMPK and autophagy | [34] |
| Oleanolic acid (3β-hydroxyolean-12-en-28-oic acid) | Different cancer model | Inducing tumor cell apoptosis via activating autophagy | [35] |
| Epigallocatechin-gallate | Human aortic epithelial cells | Autophagic flux stimulation | [36] |
| Korean red ginseng | Colon carcinoma cells (HCT-116 and SNU-1033) | Mitochondrial reactive oxygen species (ROS)-mediated autophagy and apoptosis. | [37] |
| Betulinic acid | Human malignant melanoma cell lines | Inhibits tumor growth and induces autophagy | [38] |
| Curcumin | Human thyroid cancer cells | Mitogen-activated protein kinase (MAPK) activation and mTOR inhibition with autophagy induction | [39] |
| Ursolic acid | Human breast cancer cells (MCF-7/MDA-MB-231) | Class III PI3K(VPS34)/Beclin-1 pathway autophagy induction | [40] |
| Obatoclax | Atypical teratoid/rhabdoid tumors | Dual mammalian target of rapamycin complex 1/2 (mTORC1/2) inhibition | [41] |
| Chloroquine | Triple-negative breast cancer (TNBC) | PI3K/Akt inhibitors and induces autophagy | [42] |
| Rapamycin | Kaposi’s sarcoma | PI3K/Akt/mTOR activation autophagy | [43] |
Autophagy may be tumor-suppressive, neutral, and tumor-promoting in cancer, depending on the microenvironment stress, nutrient availability, immune system, and pathogens [21]. The mTOR protein kinase is related to various biological functions, such as cell survival, growth, immunity, and metabolism. Consequently, mTOR is implicated in supervising numerous biological processes, notably autophagy, apoptosis, and the cell cycle, thus preventing the commencement of the concluding process [44]. A P75NTR activation by rapamycin induces autophagy, leading to Kaposi’s sarcoma cell death [43]. Additionally, rapamycin (sirolimus), a secondary metabolite derived from Streptomyces hygroscopicus, has significant anticancer and immunosuppressive activities [45]. Rapamycin and its semi-synthetic counterparts (rapalogs) are allosterically-precise and potent mammalian targets of rapamycin complex 1 (mTORC1) blockers that influence downstream targets, such as autophagy stimulation [46]. Other mTOR blockers interfere with adenosine triphosphate (ATP), preventing the phosphorylation of its corresponding proteins and leading to more effective mTOR suppression [47]. Altogether, these observations show that mTOR blockers may be used to promote cell death through several pathways. Their mechanisms of action rely on the tumor microenvironment, overcoming cancer cell resistance in combination therapy [48].
While cannabinoids have shown strong anticancer properties associated with autophagy, they have also shown cytoprotective properties, depending on the cell type and cannabinoid used [49]. Active cell death (ACD) in melanoma and glioma cells occurs via mTORC1 inhibition and autolysosome permeabilization, resulting in cathepsin secretion and apoptosis activation [50]. JWH-015, for example, is a synthetic cannabinoid specific to the CB2 receptor that suppressed tumor development in hepatocellular carcinoma (HCC) cells in vivo via inhibiting the Akt/mTORC1 cascade through an AMP-activated protein kinase (AMPK) stimulation [9]. Moreover, obatoclax (GX15-070) is another BH3 mimic that induced necroptosis in oral squamous cell carcinoma, acute lymphoblastic leukemia, and rhabdomyosarcoma via autophagic signaling [51]. Additionally, obatoclax promoted autophagy in adenoid cystic carcinoma cells and inhibited autophagy in colorectal cancer cells in the absence of Bcl-1 [52]. Subsequently, ABT-737 was found to be effective in vitro against HCC cells via an autophagy-dependent mechanism involving Bcl-1 [53].
Furthermore, APO866 inhibited nicotinamide adenine dinucleotide biosynthesis and induced apoptotic cell death in hematological cancer cells [54]. Elsewhere, histone deacetylase (HDAC) inhibitors were shown to induce autophagy. A recent study used the HDAC inhibitor, suberoylanilide hydroxamic acid (SAHA), to treat cutaneous T-cell lymphoma and glioblastoma [55]. While apoptosis had been identified as the primary mechanism through which the HDAC inhibitors promoted cancer cell death, autophagy promotion was also associated with a PI3K/Akt/mTOR pathway inhibition [56]. In addition, SAHA inhibited breast cancer cell proliferation in vitro by stimulating autophagy and activating cathepsin B [57,58]. Moreover, in vivo and in vitro studies have shown that MHY2256 (a synthetic class III HDAC inhibitor) stimulates CCA, ACD, and apoptosis in endocervical curettage [59]. SAHA activates autophagy by inhibiting mTOR and upregulating the LC3 expression. Autophagy mitigates a SAHA-induced apoptotic and nonapoptotic cell death, suggesting that targeting autophagy may improve SAHA’s therapeutic effects [60].
2. Targeted Drug for the Inhibition of Autophagy in Cancer Therapy
It has been shown that inhibiting autophagy makes an anticancer therapy more effective, suggesting that it is a potentially valuable approach that could be combined with other anticancer therapeutic approaches to improve cancer treatments [61]. Numerous autophagy inhibitors impede the autophagy processes at various stages, as discussed in Table 2 and Figure 4.
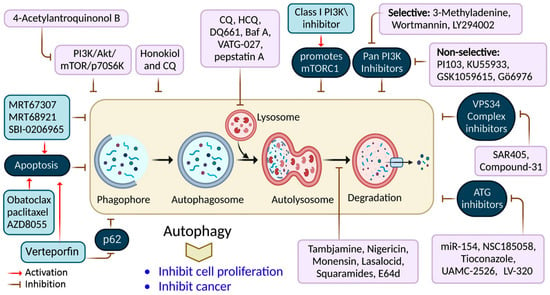
Figure 4. Autophagy inhibition by several compounds and drugs used in cancer therapy.
Table 2. Recently used autophagy inhibitors and their mechanisms of action in different cancers.
| Drug | Model | Molecular Mechanism | Reference |
|---|---|---|---|
| Nigrosporins B | Human cervical cancer cells (Ca Ski) | PI3K/AKT/mTOR-mediated autophagy inhibition | [62] |
| 3-methyladenine (3-MA) | Gastric cancer cells | PI3K/AKT/FOXO3a inhibition | [63] |
| 4-acetylantroquinonol B | Human pancreatic cancer cells (MiaPaCa-2 and GEM-resistant MiaPaCa-2) | Upregulation of reactive oxygen species (ROS) promoted apoptosis via autophagy inhibition | [64] |
| LY294002 | Laryngeal squamous cell carcinoma | Inhibition of autophagy via the PI3K/mTOR pathway | [65] |
| MRT67307 | Several cancer models and lung cancer cells (A549) | ULK1 inhibition and autophagy inhibition | [66] |
| Honokiol | Human medulloblastoma | ERK/p38-MAPK-mediated autophagic inhibition | [67] |
| Obatoclax | Different cancer cells | Mitochondrial Ca2+ overload and autophagy inhibition | [68] |
| MHY2245 | Human colorectal cancer cells (HCT116) | Cell cycle arrest and apoptosis | [69] |
| Hydroxychloroquine | Human pancreatic cancer cells (PANC-1, MiaPaCa-2, and BxPC-3) | Promotes Bcl-xL inhibition-induced apoptosis with autophagy inhibition | [70] |
| Wortmannin | Pancreatic cancer cells (PANC-1, BxPC-3, and Capan-2) | Nutrient-starvation conditions via autophagy inhibition | [71] |
| Bafilomycin A1 | Thyroid cancer cells (MDA-T32, MDA-T68, FTC-133, and 8505c) | Autophagy inhibition reduced cell migration and invasion | [72] |
| Tioconazole | Human colorectal cancer cells (HCT116) | Autophagy inhibition | [73] |
Nigrosporins B, a potential anti-cervical cancer agent, induced apoptosis and protective autophagy in human cervical cancer Ca Ski cells. These effects were mediated via the PI3K/AKT/mTOR signaling pathway [62]. Additionally, autophagy is affected by antimetabolites, antiangiogenic agents, DNA-damaging agents, proteasome inhibitors, microtubule-targeted drugs, death receptor agonists, hormonal agents, HDAC inhibitors, and kinase inhibitors [74]. ATG proteins and other factors form complexes that lengthen phagophores. ATG7 is involved in creating the ATG5-ATG12 complex and coupling PE to LC3 and GABARAP [75]. Consequently, many ATG7 suppressors (e.g., WO2018/089786) have been developed, extending the use of micro RNAs (miRNAs) targeting the ATG7 gene, including miR-154, which suppresses blade cancer growth [76]. Conversely, ATG4B cleaves LC3, stimulating it in preparation for coupling with PE, which is required for autophagosome development and activation [77].
Verteporfin is a photodynamic therapeutic agent derived from benzoporphyrin. It also suppresses autophagosome production induced by serum and glucose deprivation but not mTOR suppression [78]. Verteporfin is suspected of suppressing p62 oligomerization, a protein crucial for ubiquitinated targets to be sequestered into autophagosomes [78]. Inhibiting ULK1 slowed the tumor development and activated apoptosis. This finding has resulted in the discovery of many chemicals that compete for ATP-binding sites, including compound-6, MRT67307, and MRT68921 [9]. Apart from these, the most investigated compound is SBI-0206965, which has been shown to suppress autophagy and promotes apoptosis in neuronal ceroid lipofuscinoses, clear-cell renal cell carcinoma cells, and non-small-cell lung cancer (NSCLC) cells [79]. SF1126, a conjugated LY294002 derivative, was engineered to assemble in integrin-expressing tissues, increasing LY294002′s solubility and pharmacokinetics, promoting tumor site retention, and showing anticancer and antiangiogenic activity in mouse models [80].
3-methyladenine (3-MA) was among the earliest autophagy inhibitors. It inhibits autophagy in starvation situations by inhibiting PI3KC3 [81]. Conversely, when nutrients are present, they induce autophagy by blocking PI3KC1. The VPS34 inhibitor, SAR405, is a (2S)-tetrahydropyrimido-pyrimidinone class member that inhibits kinases via an extreme competition for their ATP binding site, thus suppressing autophagy produced by an mTOR inhibition or malnutrition [82]. VPS34-IN1 is a bipyrimidinamine that preferentially suppresses PI3KC3 [9]. Esomeprazole’s antiproliferative action, for example, was enhanced when combined with the PI3K inhibitor, 3-MA, in gastric cancer cells through EGFR downregulation via the PI3K/FOXO3a pathway [63]. In gemcitabine-resistant pancreatic cancer cells, however, 4’-acetylantroquinonol B promoted cell death and inhibited autophagy by downregulating the PI3K/Akt/MDR1 pathway, increasing cell death [64].
Autophagy’s late phase involves autophagosomes fusing with lysosomes, which contain hydrolases that destroy the autophagosomes’ contents. During this stage, autophagy is suppressed by the lysosomal blockers, bafilomycin A1 (Baf-A1), chloroquine (CQ), and its derivatives Lys05 and hydroxychloroquine (HCQ), which are antimalarial medicines used for treating malaria and, more recently, cancer [50].
Only CQ and HCQ are authorized for therapeutic use as autophagy inhibitors [42]. Consequently, CQ derivatives with increased inhibitory-efficacy against autophagy have been synthesized. Lys05, for example, is a dimeric CQ analog that aggregates better than HCQ within acidic organelles, particularly lysosomes [83]. Duroquinone DQ661 disrupts lysosomal degradation, notably autophagy, and suppresses PPT-1, inhibiting the mTORC1 cascade. Furthermore, DQ661 has shown independent efficacy in tumor mouse models and the ability to overcome gemcitabine resistance [9]. VATG-027 is another antimalaria drug that suppresses autophagy with antitumoral effects [84]. Meanwhile, mefloquine accumulates in lysosomes, impairing autophagy, inducing apoptosis, and inhibiting MDRP1, making it potent against MDRT cells. It sensitizes chronic myeloid leukemia cells created from chronic-stage patients to tyrosine kinase blockers, with a preference for stem/progenitor tumor cells over normal cells [85].
CQ and its analogs, however, are not the only medications that suppress autophagy by affecting lysosomes [86]. Baf A is a vacuolar-H+ ATPase blocker that prevents H+ from entering lysosomes, vacuoles, and vesicles. Baf A also suppresses autophagosome-lysosome fusion by disrupting the Ca2+ gradients required for this pathway [87]. Additionally, ionophores can alter the pH of the lysosome, affecting the autophagy mechanism. Tambjamine derivatives are ASI synthesized from naturally produced tambjamines [88]. Importantly, because lysosomes are involved in tumor invasion, these blockers are efficacious against metastasis by addressing cancer stem cells and promoting tumor vessel normalization.