1. Classical Mutations
Ex19Del and L858R are found in 71.9% of NSCLCs with an
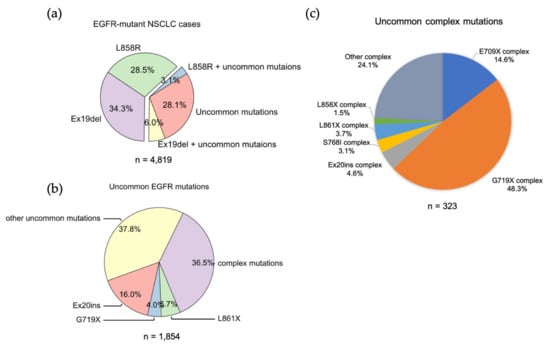
EGFR mutation. Ex19Del and L858R are the sole EGFR mutations in 34.3% and 28.5%, respectively, while co-occurrence of Ex19Del or L858R with uncommon EGFR mutations accounts for 6.0% and 3.1% of cases (
Figure 12a). Ex19Del and L858R as classical mutations confer sensitivity to all first-, second- and third-generation EGFR TKIs
[1][2][3][4][5][6][7][10,20,22,55,56,61,62] (
Figure 23), and co-occurrence of an uncommon mutation does not impact TKI sensitivity
[8][9][40,63]. In fact, patients with complex “classical” mutations (Ex19Del or L858R in complex with an uncommon EGFR mutation) might have more favorable clinical outcomes than patients with uncommon mutations alone
[10][11][64,65].
ThWe
researchers have not found any classical + classical complex mutations in
theour analysis of GENIE dataset. The sensitivity of this class of complex mutations might be equivalent to the corresponding single mutation, as L858R + Ex19Del complex mutations display similar patient outcomes with EGFR TKI treatment compared to patients with single classical mutations alone
[12][66].
Figure 12. The frequencies of EGFR mutations in NSCLC. (a) Data were acquired from GENIE dataset (GENIE Cohort v12.0-public, n = 153,834). Data were filtered to contain mutations from NSCLC (n = 22,050). The common resistance mutations T790M and C797S were filtered out. (b) Pie chart showing the frequency of different uncommon EGFR mutations in NSCLC. (c) Pie chart showing the frequency of different uncommon complex EGFR mutations in NSCLC.
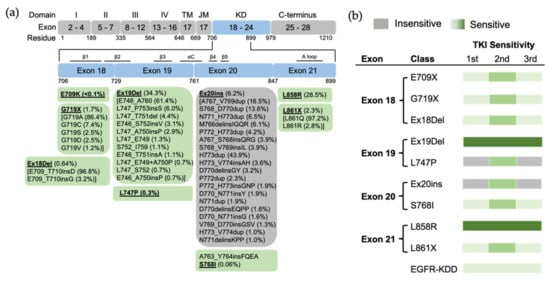
Figure 23. The position of EGFR mutations and sensitivity to EGFR TKIs based on exon-grouped classification. (a) Exons 18–21 in the tyrosine kinase region where the relevant mutations are located are expanded, and a detailed list of EGFR mutations in these exons is shown in the boxes below. TM, transmembrane; JM, juxtamembrane; KD, kinase domain. (b) The sensitivity to EGFR TKIs based on exon-grouped classification. Gray, insensitive to EGFR TKIs. Green, sensitive to EGFR TKIs in which the light-to-strong color represents weak-to-strong sensitivity.
Structurally, Leu858 lies within the two-turn helix of the A loop and packs against the αC helix in the “Src-like inactive” conformation. Arginine has a much larger side chain than leucine, and substitution of Leu858 with arginine is not tolerated with the inactive conformation
[2][20]. The positively charged Arg858 is surrounded by a cluster of negatively charged residues (Glu758, Asp855, and Asp837), which stabilize the αC helix and the KE salt bridge. Therefore, the substitution holds the kinase domain in the active conformation likely at the expense of locally disordered conformation, rather than “Src-like inactive” conformation
[13][21]. As a result, L858R mutation promotes the ligand-independent dimerization of EGFR kinase
[14][24]. Structure-based approaches classify L858R into classical-like mutations, as Arg858 is located at the N-terminal portion of the A loop in the C-lobe, which is far from the ATP-binding pocket
[15][27]. A spectrum of rare L858R + mutations were also categorized into classical-like mutations
[15][27]. This subgroup of mutations is sensitive to all first-, second- and third-generation EGFR TKIs (
Figure 34).
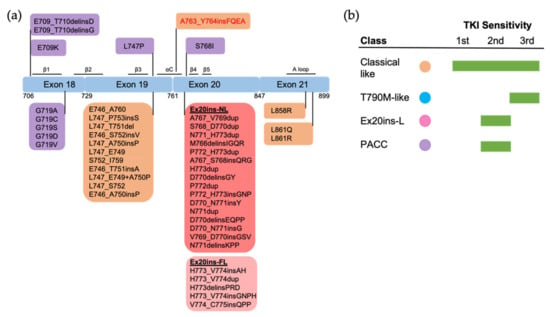
Figure 34. The positions and structure-based classification of EGFR mutations. (a) A detailed list of EGFR mutations in each exon are shown in the below boxes. Different colors of each box represent different classes of mutations following structure-based classification. Ex20ins-L, exon 20 loop insertions; Ex20ins-NL, exon 20 near-loop insertions; Ex20ins-FL, exon 20 far-loop insertions; PACC, P-loop and αC-helix compressing mutations. (b) The sensitivity to EGFR TKIs of each group of mutations based on structure-based classification.
Ex19Del mutations shorten the length of β3-αC loop and suppress the locally disordered conformation
[13][21], and therefore constrain the αC helix to the active conformation
[3][22], with the KE salt bridge making the interaction between the kinase domain and ATP favorable
[16][67]. There have been more than 20 Ex19Del variants identified in NSCLC
[17][68]. The length of the β3–αC loop functions as a rheostat for kinase activity. Specifically, deletion of five amino acids (E746_A760) makes an optimal orientation of the αC helix for catalytic activity, while other deletions also result in structural perturbations of the αC helix that impair kinase activity compared to WT EGFR
[3][22]. This conformational difference leads to variable ability to form ligand-independent dimerization and therefore differential sensitivity to EGFR TKIs
[17][18][68,69]. Clinically, variations in Ex19Del are translated into differential clinical features and treatment outcomes
[19][70]. Ex19Del mutations were classified into classical-like mutations as, structurally, the β3–αC loop in the N-lobe is far from the ATP-binding pocket
[15][27]. A spectrum of Ex19Del + rare mutations were also categorized into classical-like mutations (
Figure 34).
Ex19Del and L858R mutants have a lower competitive binding affinity for ATP than WT EGFR. First-generation EGFR TKIs bind more favorably to the kinase domain of mutants than WT, thereby inhibiting mutant kinase activities in a therapeutically useful manner
[2][20]. The substitution of Thr790 to the bulky methionine increases the ATP affinity and stabilizes the active conformation of EGFR kinase domain, which sterically prevents the binding of first-generation TKIs, thereby imparting drug resistance
[20][71]. Second-generation TKIs bind irreversibly to EGFR Cys797 at the margin of the ATP-binding cleft by covalent adduct formation, and were anticipated to overcome acquired resistance caused by T790M mutation
[21][72]. However, the poor selectivity of second-generation TKIs for T790M over WT EGFR results in dose-limiting toxicities that limit clinical efficacy
[6][61]. Osimertinib has a differential binding pose with Met790 from Thr790 of WT EGFR, thus yielding greater selectivity for T790M mutation over WT EGFR than afatinib
[22][25]. Greater selectivity has translated into more favorable treatment outcomes, as either second-line or first-line therapy, than earlier-generation EGFR TKIs
[23][24][52,53].
2. Uncommon EGFR Mutations
2.1. Exon 18 Mutations
EGFR exon 18 mutations include E709_T710insX (e.g., exon 18 deletion, Ex18Del), E709X and G719X (
Figure 12b). From the analysis of the GENIE dataset, either E709X or E709-T710insX as a single
EGFR mutation accounts for around 0.68% of all
EGFR mutations and 1.7% of rare
EGFR mutations in NSCLC (data not shown). Around 96% of E709X co-occurs with another EGFR mutation, such as G719X or L861X
(Table S3 and S9). G719X mutations as independent
EGFR mutations, including G719A, G719C, G719S, G719D and G719V, and account for 4.0% of rare
EGFR mutations and 1.6% of all
EGFR mutations in NSCLC. G719X mutations also co-occur with other
EGFR mutations, which represent 8.4% of rare
EGFR mutations and 3.2% of all
EGFR mutations in NSCLC (
Figure 12c).
Preclinical studies show that exon 18 mutations including E709K, E709_T710insD and G719A are sensitive to first-, second- and third-generation EGFR TKIs, but show less sensitivity than Ex19Del
[25][39] (
Figure 23). Exon18 mutations tend to be more sensitive to second-generation TKIs than first- and third-generation EGFR TKIs
[8][17][18][40,68,69]. E709K and G719A tend to be more sensitive to EGFR TKIs than E709-T710insD
[8][17][40,68]. Clinically, patients with G719X and Ex18Del respond poorly to first-generation TKIs
[25][39], and patients with G719X also respond poorly to the third-generation TKI osimertinib
[19][70]. Few clinical data about second- and third-generation TKIs exist for E709X as a single EGFR mutation.
Both Glu709 and Gly719 are located in the N-lobe. Glu709 is located on the N-terminal end of the β1 strand, which is immediately followed by the P loop. Gly719 is the first glycine in the evolutionarily conserved “GXGXXG” motif of the P loop. Substitution of Gly719 to non-glycine residues decreases the flexibility of the P loop and attenuates the hydrophobic interactions that constrain the αC-helix in the inactive conformation
[2][20]. Therefore, G719X mutations destabilize the inactive conformation and are well accommodated in the active conformation
[2][13][20,21]. This conformation increases the propensity for dimerization and subsequent receptor activation. Exon 18 mutations, including E709X, G719X and Ex18Del mutations, were categorized as PACC mutations
[15][27] (
Figure 34). Structurally, PACC mutations change the flexibility of the P loop and destabilize an osimertinib-binding pattern. By contrast, second-generation TKIs do not interact with the P loop and favor the binding pose. Therefore, PACC mutations are more sensitive to the second-generation TKIs than other-generation TKIs
[15][27]. Clinically, patients with G719X mutations are more responsive to second-generation EGFR TKIs than first- and third-generation TKIs
[21][26][27][42,72,73]. Similarly, patients who develop resistance to osimertinib via a mutation in Gly724, the third glycine in the “GXGXXG” motif, often respond to second-generation EGFR TKIs
[28][29][30][31][74,75,76,77], but not first-generation TKIs
[32][78]. The variable sensitivity of different exon 18 mutations observed in previous studies might be explained by the differential flexibility of the P loop induced by mutations.
2.2. Exon 19 Mutations
ThWe
researchers found 13 cases with L747P mutation in the GENIE dataset.
The researchers aWe also found 5 cases with L747X complex mutations, including L747F + L861Q (1 case), L747S + G719C (2 cases) and L747A + I744M (2 cases).
The researchers We have not found exon 19 insertions in the GENIE dataset, while this subset of mutations has been reported to account for approximately 1% of
EGFR-mutant NSCLC
[9][63]. Available clinical data indicate that L747P/S mutations are resistant to first- and third-generation EGFR TKIs
[33][34][79,80] and confer sensitivity to second-generation EGFR TKIs
[35][36][81,82]. Consistent with this evidence, L747X mutations were included in PACC mutations, as substitution of Leu747 to proline or serine might alter the rigidity of the β3–αC loop, which stabilizes the αC-helix and KE salt bridge interactions in the active conformation
[37][83].
2.3. Exon 20 Mutations
EGFR exon 20 insertion (Ex20ins) mutations contain a broad spectrum of small insertions of 1–7 amino acids that occur within the C-terminal end of the αC-helix and the immediately following loop
[38][37]. After classical mutations, Ex20ins mutations are the next most prevalent
EGFR mutation in NSCLC
[39][84]. From the GENIE dataset,
thwe
researchers found more than 60 different forms of Ex20ins mutations, accounting for 16% of rare
EGFR mutations and 6.2% of all
EGFR mutations in NSCLC (
Figure 12).
ThWe
researchers aalso found other forms of exon 20 mutations, including S768I (3 cases), V774L (1 case) and Q787L (1 case) (
Figure 23). Exon 20 mutations also co-occur with other
EGFR mutations (
Figure 12c).
Variable TKI sensitivity has been found in different types of Ex20ins mutations
[40][41][85,86]. Most patients harboring Ex20ins mutations are insensitive to most of the FDA-approved EGFR TKIs
[42][43][44][45][87,88,89,90]. However, a specific subset of insertions within the C-terminal portion of the αC-helix, for example, A763_Y764insFQEA, are sensitive to first-generation EGFR TKIs
[46][44]. Multiple reports have demonstrated A763_Y764insFQEA is also responsive to second- and third-generation EGFR TKIs with comparable sensitivity to classical mutations
[44][47][48][45,89,91]. For the rest of the Ex20ins mutations, most of them do not respond to first- and third-generation TKIs
[49][41], but are sensitive to second-generation TKIs
[50][92]. Exceptionally, the three insertion mutations (D770_N771insG, D770>GY, and N771_P772insN) are at least partially responsive to first-generation EGFR TKIs
[49][41].
Structural variations in Ex20ins mutations lead to the heterogeneity of TKI sensitivity. The insertions form a wedge that “pushes” the αC-helix and hold the αC in the active conformation
[46][51][23,44], leading to constitutive activation of the receptor
[46][51][23,44]. Sequence and conformational variation in the αC–β4 loop may function as a rheostat that regulates the kinase activity via modulating the αC-helix conformation
[51][23]. Exceptionally, the insertion A763_764insFQEA that occurs within the αC-helix differs from insertions within the αC–β4 loop, and may constitutively activate the receptor most similarly to classical mutations, rather than other Ex20ins mutations. As might be expected, A763_764insFQEA is sensitive to first-, second- and third-generation EGFR TKIs
[44][47][48][45,89,91], and is classified as a classical-like mutation. Other Ex20ins mutations are categorized as a unique class called Ex20-L mutations
[15][27]. Ex20ins-L are sensitive only to select second-generation TKIs such as poziotinib
[52][93], and Ex20ins-active TKIs such as CLN-081
[53][94] and mobocertinib
[54][95]. Ex20ins-L mutations could be further subdivided into Ex20ins near αC-β4 loop (Ex20ins-NL) and far αC–β4 loop insertions (Ex20ins-FL). Ex20ins-NL is more sensitive to second-generation and Ex20ins-active TKIs than Ex20ins-FL (
Figure 34).
Patients with single S768I mutation have variable responses to first-generation EGFR TKIs
[45][55][56][57][90,96,97,98]. Patients whose cancers harbor theS768I mutation responded to afatinib
[42][87], leading to FDA approval of this drug for this subset of patients. Notably, S768I usually co-occurs with additional
EGFR mutations (7/8 cases in this study), making the response of the single S768I mutation to afatinib ambiguous. Co-occurrence of S768I with additional
EGFR mutations might impact TKI sensitivity
[58][59][99,100]. One patient with S768I+V769L, for example, was resistant to afatinib
[60][101]. S768I-complex mutations are sensitive to the third-generation EGFR TKI osimertinib
[61][62][102,103], though little data exists on single-mutant S768I sensitivity. S768I and its complex mutations are classified as PACC mutations. Ser768 is located at the αC–β4 loop. Substitution of Ser768 to isoline improves the hydrophobic interactions between the αC helix and the adjacent β9 strand, which might enthalpically stabilize the “αC-in” active conformation
[13][21]. As such, S768I and its complex mutations are classified as PACC mutations (
Figure 34).
Finally, little clinical or structural data exist for V774L and Q787L, though V774M and its complex mutations are classified as PACC mutations.
2.4. Exon 21 Mutations
Apart from L858R mutation,
thwe
researchers found another exon 21 mutation L861X, including L861Q and L861R, in 2.3% of all
EGFR mutations and 5.7% of rare
EGFR mutations in the GENIE lung cancer dataset (
Figure 12b). L861X mutations were also found to co-occur with additional
EGFR mutations (
Figure 12c). Preclinical and clinical studies show that L861Q mutation has an intermediate sensitivity to first-generation EGFR TKIs comparable to S768I and G719X mutations
[26][63][64][42,43,104] (
Figure 23). Patients with L861Q mutation are responsive to the second-generation EGFR TKI afatinib
[42][87], which led to FDA approval of afatinib for this subtype
[1][10]. In preclinical studies, L861Q mutations are sensitive to the third-generation EGFR TKI osimertinib
[63][43], and a phase II trial reported patients with L861Q having partial responses to osimertinib
[61][102]. Structurally, substitution of Leu861 to Gln allows the formation of new hydrogen bonds near the C-terminal of the αC helix that might enthalpically stabilize the “αC-in” active conformation
[13][21]. L861X mutation and its complex mutations have been classified as PACC mutations (
Figure 34).
2.5. EGFR Kinase Domain Duplication
In-frame, tandem duplication of
EGFR exons 18–25, encoding EGFR kinase domain duplication (EGFR-KDD), has been identified in patients with NSCLC
[65][105], with cases of a duplication of exons 14–26, 17–25 and 18–26 also reported in NSCLC
[66][106]. EGFR-KDD occurs in 0.2–0.24% of all
EGFR-mutant NSCLC patients
[66][67][68][106,107,108]. Current evidence suggests that EGFR-KDD is sensitive to multiple EGFR TKIs
[65][66][105,106]. Case studies show that NSCLC patients with EGFR-KDD can have durable partial response to either first-line
[66][106] or second-line treatment with gefitinib
[69][109], though at least one patient with EGFR-KDD did to respond to first-line gefitinib
[66][106]. Case studies also show that NSCLC patients with EGFR-KDD are at least partially responsive to afatinib treatment
[65][70][105,110], and the third-generation TKI osimertinib has been found to be effective in patients with EGFR-KDD
[71][72][111,112].
Structure–function studies indicate that EGFR-KDD can form ligand-independent intramolecular asymmetric dimers in which one kinase domain functions as “activator” (donor) and the other functions as “receiver” (acceptor), and ligand-dependent intermolecular asymmetric dimers and higher-order oligomer. The linker between the two kinase domains provides additional enthalpic stabilization to promote activation. Preclinical studies show that the inhibition of EGFR-KDD activity can be achieved by the inhibition of intramolecular kinase activity (afatinib) and intermolecular kinase activity (cetuximab)
[73][48]. Compared to gefitinib, structural studies predict osimertinib to bind more favorably with, and thus better inhibit, EGFR-KDD
[74][113]. More structure–function studies are required to understand the detailed conformations when EGFR-KDD binds to the available TKIs, so that
thwe
researchers ccan classify this type of mutations based on the structure-based grouping.
2.6. Uncommon Complex Mutations
ThWe researchers found uncommon complex mutations (rare + rare) occur in 36.5% of rare EGFR mutations and 6.7% of all EGFR mutations in the GENIE lung cancer dataset (Figure 12C). Of them, G719X complex mutations are the most common subtype of mutations and account for almost a half of all uncommon complex mutations (Figure 12C), with G719X + S768I most common one (Table 12). E709X complex mutations are the second most common of this subtype of mutations and account for 14.6% of all uncommon complex mutations (Figure 12C). Interestingly, all E709X mutations co-occur with G719X mutations (Table 12). ThWe researchers aalso found Ex20ins, L861X, L858X and S768I complex mutations (Figure 12C, Table 23).
Table 12.
Complex mutations identified in exon 18 from GENIE lung cancer dataset.
Table 23.
Complex mutations identified in exon 20 and 21 from GENIE lung cancer dataset.
Uncommon
EGFR complex mutations encompass a wide range of patient responses to EGFR TKIs. Structure-based approaches classify the majority of uncommon EGFR complex mutations as PACC mutations, including E709X + G719X, G719X + rare and S768I + rare, like their single mutation partners. These complex mutations tend to yield more favorable patient outcomes in response to TKIs than the single rare mutations alone
[43][64][75][76][88,104,114,115]. Interestingly, the sensitivity to EGFR TKIs is likely influenced by the specific co-occurring partner mutation. For example, patients with G719X + S768I mutations have dramatically different clinical outcomes compared to patients with G719X+L861Q mutations
[64][104]. Single S768I mutation might not be sensitive to erlotinib, but co-occurrence with sensitizing
EGFR mutations will confer sensitivity to the single S768I mutation
[58][99].